Automated Fast-Flow Synthesis of Peptide-PNA Conjugates: A Transformative Platform for Accelerated Therapeutic Development
The therapeutic potential of peptide nucleic acids (PNAs) is often limited by challenging synthesis and poor cellular delivery.
Automated Fast-Flow Synthesis of Peptide-PNA Conjugates: A Transformative Platform for Accelerated Therapeutic Development
Abstract
The therapeutic potential of peptide nucleic acids (PNAs) is often limited by challenging synthesis and poor cellular delivery. This article explores the emergence of automated, high-temperature fast-flow synthesizers as a transformative solution, enabling the rapid and efficient production of peptide-PNA conjugates (PPNAs). We cover the foundational principles of PNA technology and its inherent challenges, detail the engineering and operation of automated flow platforms, provide strategies for optimizing synthesis and enhancing bioavailability, and validate the platform's utility through compelling biological applications in antivirals and antibacterials. This overview is tailored for researchers, scientists, and drug development professionals seeking to leverage this efficient technology for advanced antisense therapy development.
Peptide Nucleic Acids (PNAs): Unlocking Potential Through Synthesis and Delivery
This application note details the superior biophysical properties of Peptide Nucleic Acids (PNAs), focusing on their enhanced stability and binding affinity compared to natural nucleic acids. PNAs, with their unique N-(2-aminoethyl)glycine backbone, exhibit remarkable resistance to enzymatic degradation and form more stable duplexes with complementary DNA and RNA. Framed within research on automated synthesis platforms, this document provides validated protocols and quantitative data to support the integration of PNA conjugates into modern drug discovery pipelines, offering researchers a practical guide to leveraging these advanced biomolecules.
Peptide Nucleic Acids (PNAs) are synthetic nucleic acid analogs in which the native sugar-phosphodiester backbone is replaced by an uncharged, achiral backbone composed of repeating N-(2-aminoethyl)glycine units [1] [2]. This fundamental structural modification confers several superior properties over natural DNA and RNA. The neutral polyamide backbone eliminates electrostatic repulsion with complementary nucleic acid strands, leading to significantly higher binding affinity and sequence specificity [1]. Furthermore, the PNA backbone is resistant to degradation by both nucleases and proteases, granting it exceptional metabolic stability in biological environments, a critical advantage for therapeutic applications [1] [2].
The following diagram illustrates the core structure and binding modes of PNA compared to DNA.
PNA's synthetic nature allows for extensive backbone and nucleobase modifications, enabling fine-tuning of properties like solubility, specificity, and cellular uptake [1] [3]. These characteristics make PNA a powerful tool in antisense therapy, molecular diagnostics, and biotechnology. The recent development of automated, high-throughput synthesis platforms for peptide-PNA conjugates (PPNAs) has further accelerated their application in drug discovery and nanotechnology, making robust protocols for their production and evaluation more essential than ever [4] [5].
Quantitative Analysis: PNA vs. Natural Nucleic Acids
The superior performance of PNA is quantitatively demonstrated through its enhanced thermal stability and binding properties. The data below compare PNA-DNA duplexes with natural nucleic acid duplexes.
Table 1: Comparative Thermal Stability of Nucleic Acid Duplexes (15-mer sequences)
| Duplex Type | Average Melting Temperature (Tm) | Effect of Single Mismatch (ΔTm) | Ionic Strength Dependence |
|---|---|---|---|
| PNA-DNA | ~70 °C [1] | ~ -15 °C [2] | Low [2] |
| DNA-DNA | ~55 °C [1] | ~ -11 °C [2] | High |
| PNA-RNA | Higher than DNA-RNA [5] | More destabilizing [2] | Low |
The elevated melting temperature of PNA-DNA duplexes, a direct result of the lack of inter-strand electrostatic repulsion, underscores PNA's higher binding affinity [1]. The greater destabilization caused by a single base mismatch highlights its exceptional sequence specificity, a crucial feature for diagnostic applications and therapeutic target engagement [2].
Recent advances in synthesis have enabled the production of complex PNA architectures with optimized properties. For instance, automated flow synthesis has been used to produce cyclic PNA and PPNA structures with 2- to 12-residue sequences achieving up to 95% crude purity, while also demonstrating remarkable nuclease resistance and permeability [4] [6].
Experimental Protocols
Protocol 1: Automated Fast-Flow Synthesis of PNA Conjugates
This protocol describes the automated synthesis of peptide-PNA conjugates (PPNAs) using a fast-flow instrument, reducing coupling times to seconds and improving crude purity [5].
- Objective: To synthesize PPNA conjugates in a single, fully automated run with high efficiency and purity.
- Principle: The method utilizes solid-phase synthesis with a flow-based instrument. High-temperature flow chemistry (70 °C) accelerates amide bond formation and reduces on-resin aggregation, enabling the synthesis of longer sequences without capping steps [5].
Materials & Reagents
- Resin: Rink Amide resin (0.5 mmol/g) [5].
- Monomers: Fmoc/Bhoc protected PNA monomers [5] [7].
- Activator: PyAOP ((7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate) [5].
- Base: DIEA (N,N-Diisopropylethylamine) [5].
- Solvent: Anhydrous DMF (Dimethylformamide) [5].
- Deprotection Reagent: 20% (v/v) Piperidine in DMF [5].
Procedure
- Reactor Setup: Load 15 mg of Rink Amide resin (0.5 mmol/g) into the reusable reactor body for a 7.5 μmol scale synthesis [5].
- Coupling Cycle:
- Deprotection: Piperidine solution flows through the reactor to remove the Fmoc protecting group. The temperature is maintained at approximately 40 °C for this step to minimize nucleobase adduct formation [5].
- Repetition: Steps 2 and 3 are repeated for each monomer addition. The entire synthesis cycle per residue is completed within 3 minutes [5].
- Cleavage & Purification: After sequence assembly, the crude PPNA is cleaved from the resin using a TFA-based cocktail, precipitated, and purified via reverse-phase HPLC [5] [3].
Protocol 2: Evaluating Cellular Delivery with CPP-PNA Complexes
This protocol outlines a method for forming non-covalent complexes between PNA and cell-penetrating peptides (CPPs) to evaluate and optimize cellular delivery in vitro [7].
- Objective: To establish and optimize non-covalent PNA-CPP complexes for efficient delivery of PNAs to the cell nucleus.
- Principle: Cationic CPPs form complexes with neutral PNA molecules through electrostatic and hydrophobic interactions, facilitating uptake across cell membranes. Incorporating a nuclear localization signal (NLS) further enhances nuclear delivery [7].
Materials & Reagents
- PNA: Synthesized PNA with a C-terminal NLS sequence (e.g., Pro-Lys-Lys-Lys-Arg-Lys-Val-amide) [7].
- CPPs: TP10, Tat, or TD2.2, each containing an NLS fragment [7].
- Cell Line: Adherent cell line relevant to the study (e.g., human ovarian epithelial cancer cell line SKOV3 [1]).
- Staining Solution: NHS-fluorescein for PNA labeling [7].
- Assay Kits: MTT cell viability assay kit.
Procedure
- Complex Formation:
- Prepare stock solutions of PNA and each CPP (TP10, Tat, TD2.2) in an appropriate buffer (e.g., water or PBS).
- Mix PNA and CPP at various molar ratios (e.g., 1:1 to 1:20) and incubate for 15-30 minutes at room temperature to allow complex formation [7].
- Cell Seeding and Treatment:
- Seed cells in a 24-well or 96-well plate and culture until they reach 60-80% confluency.
- Replace the medium with fresh medium containing the pre-formed PNA-CPP complexes. Include untreated cells as a control.
- Incubation and Analysis:
- Incubate cells for a predetermined period (e.g., 4-24 hours) at 37°C and 5% CO₂.
- Efficiency Analysis: For fluorescently labeled PNA, analyze cellular uptake using flow cytometry (e.g., FACS) and confocal microscopy [7] [3].
- Viability Assessment: Perform an MTT assay according to the manufacturer's instructions to assess potential cytotoxicity of the complexes [7].
- Optimization: Compare delivery efficiency of different CPPs and molar ratios to identify the optimal conditions, typically indicated by high fluorescence intensity (efficient delivery) and high cell viability.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for PNA Synthesis and Application
| Reagent / Material | Function / Application | Examples & Notes |
|---|---|---|
| Fmoc/Bhoc PNA Monomers | Building blocks for solid-phase PNA synthesis. | Commercially available from suppliers like Panagene; Bhoc protects the nucleobase [7]. |
| Diaminonicotinic Acid (DAN) Linker | Enables on-resin head-to-tail cyclization for cyclic PNA/PPNA synthesis [4]. | Critical for constructing cyclic architectures with improved stability and permeability. |
| Cell-Penetrating Peptides (CPPs) | Enhance cellular uptake of PNA conjugates. | TP10, Tat, TD2.2; often include an NLS for nuclear delivery [7]. |
| PyAOP / HATU | Coupling agents for efficient amide bond formation during synthesis. | Used in automated flow and manual synthesis protocols [5] [3]. |
| Cyclopentane-Modified Monomers | Backbone modification to enhance brightness in FIT-PNA RNA sensors and improve binding [3]. | e.g., cpG; introduces conformational constraint. |
| Forced Intercalation (FIT) Probes | Surrogate bases (e.g., BisQ) used in FIT-PNA for fluorescent RNA detection [3]. | Fluorescence increases upon hybridization with complementary RNA target. |
| Acetyl-pepstatin | Acetyl-pepstatin, CAS:56093-98-2, MF:C31H57N5O9, MW:643.8 g/mol | Chemical Reagent |
| Decanoyl-RVKR-CMK | Decanoyl-RVKR-CMK, MF:C34H66ClN11O5, MW:744.4 g/mol | Chemical Reagent |
Application Workflow: From Design to Functional Analysis
The following diagram summarizes the integrated workflow for developing and evaluating a functional PNA-based molecule, from design through synthesis to activity validation.
This end-to-end workflow, powered by automated synthesis, enables the rapid generation and testing of PNA constructs, significantly accelerating the development cycle for research and therapeutic applications [4] [5].
Peptide Nucleic Acids (PNAs) are synthetic oligonucleotide mimics where the sugar-phosphate backbone is replaced by an uncharged N-(2-aminoethyl)glycine backbone [8] [1]. This structure confers superior binding affinity and specificity for complementary DNA and RNA, along with high metabolic stability against nucleases and proteases [8] [9]. Despite these advantages, the translation of PNA technology into clinical and widespread practical use has been hampered by three persistent challenges: inherently poor aqueous solubility, limited cellular permeability, and difficulties in synthesizing certain sequences [8] [9] [10]. This application note details these hurdles and frames them within the context of modern solutions, particularly automated synthesis platforms for peptide-PNA conjugates (PPNAs), which are pivotal for advancing PNA-based research and drug development.
The Solubility Challenge
The neutral and relatively hydrophobic nature of the PNA backbone significantly limits its solubility in aqueous solutions, a problem exacerbated for purine-rich sequences, particularly those with consecutive guanines (G) [8] [11]. This can lead to molecular aggregation and handling issues during synthesis and application [10] [12].
Strategies for Solubility Enhancement
- Terminal Modifications: Incorporating a lysine residue at the C-terminus (or both termini) introduces a positive charge, improving water solubility without significantly affecting hybridization properties [8] [12].
- Chemical Linkers: Incorporating hydrophilic spacers, such as the AEEA linker (2-(2-aminoethoxy)ethoxy acetic acid), between the PNA sequence and any conjugated moiety (e.g., a peptide or label) can dramatically improve solubility [11] [12].
- Backbone Engineering: Incorporating cationic groups into the PNA backbone itself, or using γ-substituted chiral PNA monomers, can minimize self-aggregation and enhance water solubility [1] [10].
Table 1: Strategies to Overcome PNA Solubility and Permeability Challenges
| Challenge | Specific Issue | Proposed Solution | Mechanism of Action | Key Benefit(s) |
|---|---|---|---|---|
| Poor Solubility | General hydrophobic aggregation | C-terminal Lysine | Introduces a positive charge | Electrostatic stabilization in aqueous buffer [8] [12] |
| Purine-rich sequence aggregation | Incorporation of AEEA Spacers | Increases hydrophilicity and steric separation | Reduces intermolecular stacking and aggregation [11] [12] | |
| Folding & compact structures | γ-PNA Backbone Modification | Imparts conformational control and introduces chirality | Minimizes self-aggregation; enhances binding affinity and specificity [1] [10] | |
| Cellular Permeability | Poor membrane penetration | Conjugation to Cell-Penetrating Peptides (CPPs) | Utilizes energy-dependent/independent translocation mechanisms | Actively facilitates cellular uptake across lipid bilayers [9] [5] |
| Negatively charged targets | Use of Cationic CPPs (e.g., (KFF)3K) | Promotes electrostatic interaction with anionic cell surfaces | Enhances initial adhesion and concentration at membrane interface [9] | |
| Endosomal entrapment | Exploration of novel cyclic PPNAs | Alters internalization mechanism and intracellular trafficking | Potential for improved endosomal escape and bioavailability [4] |
The Cellular Permeability Hurdle
A major bottleneck for in vivo PNA applications is poor cellular uptake. Unmodified PNAs are not efficiently internalized by eukaryotic cells and are rapidly cleared from the body, primarily through renal excretion, with a half-life as short as 17 minutes in rats [8] [9].
Conjugation as the Primary Strategy
The most prevalent and successful strategy to overcome the delivery barrier is the covalent conjugation of PNA to carrier molecules, with Cell-Penetrating Peptides (CPPs) being the most prominent [9] [5]. CPPs are short, often cationic, peptides that facilitate transport across cellular membranes.
Table 2: Efficacy of CPP-PNA Conjugates in Antimicrobial Applications
| CPP-PNA Conjugate | PNA Target Gene | Target Organism | Minimal Inhibitory Concentration (MIC) | Reference |
|---|---|---|---|---|
| (KFF)3K-PNA | acpP |
E. coli K-12 | 0.5 - 1 µM | [9] |
| (KFF)3K-PNA | acpP |
E. coli MDR | 25 µM | [9] |
| (KFF)3K-PNA | acpP |
K. pneumoniae ATCC 13883 | 2 µM | [9] |
| (KFF)3K-PNA | rpoA |
L. monocytogenes clinical isolates | 2 - 32 µM | [9] |
| (KFF)3K-PNA | gyrA |
S. pyogenes M49 | 10 µM | [9] |
Troubles in PNA Synthesis
PNA oligomers are synthesized using Solid-Phase Peptide Synthesis (SPPS) methodologies [10]. However, the process is often more challenging than conventional peptide synthesis due to the poor solubility of monomers, and the tendency for the growing PNA chain to form intra- and inter-chain aggregates on the resin, leading to inefficient coupling and low yields of full-length product [10] [11]. These issues are particularly pronounced for longer sequences (>15-mers) and purine-rich sequences [5] [12].
Automated High-Temperature Flow Synthesis: A Modern Solution
Recent advances in automated flow synthesis present a robust solution to these synthetic challenges [4] [5]. This technology platform offers several critical advantages over traditional batch synthesis methods:
- Dramatically Reduced Coupling Times: Each amide bond formation is completed in approximately 10 seconds [5].
- High-Temperature Operation: Synthesis is performed at elevated temperatures (e.g., 70°C), which reduces on-resin aggregation and side reactions, improving crude purity [5].
- Single-Shot Conjugation: The high coupling efficiency enables the direct, fully automated synthesis of long PNA sequences conjugated to peptides (PPNAs) in a single, uninterrupted run [4] [5].
- Rapid Production: This platform can synthesize a library of PPNAs in a single day, significantly accelerating structure-activity relationship studies [5].
Integrated Experimental Protocols
Protocol: Automated Flow Synthesis of PPNA Conjugates
This protocol is adapted from the automated high-purity fast-flow synthesis method for cyclic peptide-PNA conjugates [4] and the synthesis of anti-SARS-CoV-2 PPNAs [5].
Resin Preparation
- Solid Support: Use a Rink Amide Am resin (e.g., XAL-PEG-PS or PAL-PEG-PS) with a loading capacity of ~0.5 mmol/g.
- Reactor Loading: Load 15 mg of resin into a dedicated flow reactor designed for a 7.5 μmol scale synthesis.
Synthesis Cycle Programming The automated flow synthesizer should be programmed to execute the following cycle for each monomer addition, with a total cycle time of approximately 3 minutes:
- Deprotection: Pump 20% (v/v) piperidine in DMF through the reactor at a flow rate of 2.5 mL/min for 1 minute, maintaining the reactor temperature at 40°C.
- Washing: Flush the reactor with dry DMF or NMP.
- Coupling:
- Activation: Merge separate streams of 0.2 M PNA monomer in NMP, 0.2 M activator (e.g., HATU or PyAOP) in DMF, and 0.6 M base (e.g., DIEA) in DMF.
- The merged mixture passes through a heating loop at 70°C to form the activated ester.
- Reaction: The activated ester solution flows through the resin-packed reactor, maintained at 70°C, for 10 seconds to complete the coupling.
- Washing: Flush the reactor with solvent to remove excess reagents. Note: The high efficiency of this process typically eliminates the need for a capping step [5].
Final Cleavage and Deprotection
- Cleave the crude PPNA from the resin and remove permanent protecting groups (e.g., Bhoc on nucleobases) by treating with a cocktail of TFA:m-cresol (95:5) for 5-90 minutes (time depends on the resin linker).
- Precipitate the product in cold diethyl ether.
- Purify the PPNA by reverse-phase HPLC (C18 column, 0.1% TFA in water/acetonitrile gradient) and characterize by LC-MS.
Protocol: Evaluating Cellular Permeability and Efficacy
Parallel Artificial Membrane Permeability Assay (PAMPA)
- Purpose: A high-throughput method to screen the passive permeability of synthesized PNA/PPNA libraries [4].
- Procedure:
- Dilute test compounds in a pH 7.4 buffer.
- Add the donor solution to the membrane filter, which is coated with a phospholipid mixture mimicking the cell membrane.
- Place the donor plate over an acceptor plate containing blank buffer.
- Incubate the assembly for several hours.
- Analyze the concentration of compound in the acceptor well using UV spectroscopy or HPLC to calculate the apparent permeability (Papp).
Cell-Based Splice-Correction Assay
- Purpose: To functionally validate the intracellular activity and delivery efficiency of antisense PPNAs [5].
- Procedure (e.g., using an Enhanced Green Fluorescence Protein (EGFP) reporter system):
- Transfert cells (e.g., HeLa) with a reporter gene (e.g., β-thalassemia IVS2-654 mutant) harboring a mutated intron that causes incorrect splicing and loss of EGFP fluorescence.
- Treat the cells with the test PPNA, which is designed to bind the mutant splice site and restore correct splicing.
- Include controls: untreated cells and transfected PNA (without CPP) complexed with a standard transfection reagent.
- After 24-48 hours, measure the fluorescence intensity. A successful, cell-permeant PPNA will produce a dose-dependent increase in EGFP fluorescence, indicating successful internalization, correct target engagement, and functional splice correction.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PNA Synthesis and Conjugation
| Item | Function in Research | Specific Example / Note |
|---|---|---|
| Fmoc/Bhoc PNA Monomers | Building blocks for solid-phase synthesis using Fmoc chemistry. Most common and compatible with DNA/peptide synthesizers [11] [12]. | A, C, G, T monomers (e.g., LK5001-LK5004). |
| AEEA Spacer (LK5005) | Hydrophilic linker to improve solubility and conjugation efficiency between PNA and peptides/labels [11] [12]. | Essential for N-terminal labelling. |
| Cell-Penetrating Peptides | Carrier molecules covalently conjugated to PNA to enable cellular uptake. | (KFF)3K; various cationic and amphipathic peptides [9] [5]. |
| Activators / Coupling Reagents | To activate the PNA monomer's carboxylic acid for amide bond formation with the growing chain. | HATU, PyAOP, PyBOP [5] [12]. |
| Orthogonally Protected Lysine | Allows for site-specific attachment of multiple labels or functional groups via its side chain. | Fmoc-Lys(Dde)-OH or Fmoc-Lys(Mtt)-OH for selective deprotection [11]. |
| Rink Amide Resin (PAL/XAL) | Solid support that yields a C-terminal carboxamide upon cleavage. Low loading (<0.2 mmol/g) is recommended to avoid aggregation [12]. | XAL-PEG-PS enables faster cleavage (~5 min) [12]. |
| Cleavage Cocktail | To simultaneously cleave the PNA from the resin and remove permanent base protections (Bhoc). | TFA with scavengers (e.g., 5% m-cresol) [12]. |
| Izumenolide | Izumenolide, MF:C40H74O14S3, MW:875.2 g/mol | Chemical Reagent |
| Griselimycin | Griselimycin, MF:C57H96N10O12, MW:1113.4 g/mol | Chemical Reagent |
Strategic Workflow and Visualization
The following diagram illustrates the integrated strategic workflow for overcoming the key hurdles in PNA therapeutic development, from design and synthesis to functional validation.
Diagram 1: Integrated strategy for PNA development
The synthesis of these complex molecules on an automated platform can be visualized as a highly efficient, sequential process, as shown in the workflow below.
Diagram 2: Automated flow synthesis workflow
The hurdles of solubility, permeability, and synthesis are significant but surmountable. Strategic molecular design, conjugation to carrier peptides, and leveraging cutting-edge automated synthesis platforms provide a cohesive and powerful strategy to overcome these challenges. The protocols and data summarized herein provide a roadmap for researchers to efficiently produce and test functional PNA conjugates, thereby accelerating the development of PNA-based therapeutics and diagnostics.
The efficacy of many modern therapeutic and diagnostic agents is fundamentally constrained by the impermeable nature of the cellular membrane. This barrier poses a significant challenge for the intracellular delivery of macromolecular cargoes such as peptides, proteins, and nucleic acids. Cell-penetrating peptides (CPPs) have emerged as a powerful solution to this delivery imperative. These short peptides (typically fewer than 30 amino acids) facilitate the cellular uptake of diverse cargoes, ranging from small chemical compounds to large fragments of DNA, through covalent linkage or non-covalent interactions [13] [14]. Their ability to operate in a often receptor- and energy-independent manner makes them exceptionally versatile tools [14]. The integration of CPP functionality into therapeutic and diagnostic agents is particularly relevant within the context of automated platforms for peptide-PNA (Peptide Nucleic Acid) conjugate synthesis. This document outlines the core principles of CPPs, provides detailed protocols for their application, and explores their use in advanced, automated research workflows.
CPP Fundamentals and Classification
Cell-penetrating peptides are categorized based on their physical and chemical properties, which directly influence their interaction with biological membranes and their mechanism of internalization. The table below summarizes the primary classes of CPPs.
Table 1: Classification of Cell-Penetrating Peptides
| Class | Key Characteristics | Example Sequences | Origin/Type |
|---|---|---|---|
| Cationic | Rich in positively charged amino acids (e.g., Arg, Lys); strong affinity for negatively charged membrane components [13] [15]. | TAT (GRKKRRQRRRPPQ) [15], Polyarginine (Rn) [15], Penetratin (RQIKIWFQNRRMKWKK) [15] | HIV-1 TAT protein [13], Synthetic [15], Antennapedia protein [15] |
| Amphipathic | Contain alternating polar/charged and non-polar/hydrophobic residues; can form secondary structures [13] [15]. | MAP (KLALKLALKALKAALKLA) [15], Transportan (GWTLNSAGYLLGKINLKALAALAKKIL) [15], CADY (GLWRALWRLLRSLWRLLWRA) [13] | Model Amphipathic Peptide [15], Chimeric (Galanin–Mastoparan) [15], ppTG11 derivative [13] |
| Hydrophobic | Contain only apolar residues or crucial hydrophobic groups; low net charge [13]. | Information not specific in results | Synthetic/Natural [13] |
The cellular uptake of CPPs occurs through multiple, often concurrent, pathways. The precise mechanism can depend on factors such as the peptide's sequence, the type of cargo, and the target cell [13] [16]. The main mechanisms are categorized into two broad groups: endocytosis-mediated entry and direct penetration [16].
Application Note: CPPs for Nucleic Acid Delivery
Nucleic acid-based therapeutics, including siRNA, antisense oligonucleotides, and plasmid DNA, represent a promising frontier for treating genetic disorders, cancer, and viral infections. However, their high molecular weight and negative charge impede efficient cellular uptake [13]. CPPs overcome this barrier, facilitating the intracellular delivery of these macromolecules.
Key Considerations for CPP-Nucleic Acid Formulations
The formation of CPP-nucleic acid complexes can be achieved through two primary strategies:
- Covalent Conjugation: The cargo is linked to the CPP via stable (e.g., amide, thiazolidine) or cleavable (e.g., disulfide) bonds. While disulfide linkages are common due to their susceptibility to cleavage in the reductive intracellular environment, a concern is that the covalent bond may alter the biological activity of the nucleic acid [13].
- Non-Covalent Complexation: This strategy uses electrostatic and hydrophobic interactions to form nanoparticles. Short amphipathic CPPs like MPG and Pep-1 are effective carriers for this purpose. A significant advantage is that cargoes can be delivered efficiently while maintaining full biological activity, and this method avoids the need for chemical modification of the cargo [13].
For siRNA delivery, non-covalent strategies have demonstrated a more significant biological response. For instance, MPG/siRNA complexes form stable nanoparticles that protect the siRNA, show a low degradation rate, and can be easily functionalized for specific targeting. This system has been successfully used both in cultured cells and in vivo in mouse blastocytes for robust gene regulation [13].
Table 2: CPP Applications in Nucleic Acid Delivery
| Cargo Type | CPP/Delivery System | Conjugation Method | Key Application & Outcome |
|---|---|---|---|
| siRNA | Transportan, Penetratin | Covalent (disulfide linkage) | Delivery of siRNA targeting luciferase or eGFP reporters [13]. |
| siRNA | MPG | Non-covalent complexation | Efficient gene silencing in vitro and in vivo; high stability and low degradation rate [13]. |
| siRNA | CADY | Non-covalent complexation | Secondary amphipathic peptide based on tryptophan and arginine; effective siRNA delivery [13]. |
| Plasmid DNA, Oligonucleotides | S413-PV | Not specified | Exploration for intracellular delivery of nucleic acids in a therapeutic context [16]. |
Experimental Protocols
Protocol 1: Automated Solid-Phase Synthesis of a Cationic CPP (e.g., TAT)
This protocol is adapted for an automated programmable platform like the SPPS Chemputer, which uses the Chemical Description Language (χDL) to digitize and execute synthetic procedures [17].
1. Resin Swelling
- Material: Appropriate solid support (e.g., PAL-PEG-PS or XAL-PEG-PS resin, load <0.2 mmol/g).
- Procedure: In the SPPS reactor, swell the resin in a suitable solvent (e.g., DMF) for 30-60 minutes under gentle nitrogen bubbling [17] [12].
2. Peptide Chain Assembly (Repeat for each amino acid)
- Fmoc Deprotection: Treat the resin with 20% piperidine in DMF (e.g., 2 x 9 mL, 15 min total). This can be monitored in real-time via UV monitoring to verify completion [17] [18] [19].
- Washing: Wash the resin multiple times with DMF.
- Amino Acid Coupling:
- Activation: Pre-activate the Fmoc-protected amino acid (0.5 M in DMF) with HATU (0.45 M in DMF) and DIPEA for a set time (e.g., 2.5 min).
- Coupling: Transfer the activated amino acid to the SPPS reactor and couple for a defined period (e.g., 1 x 30 min). Induction heating can be applied to accelerate coupling and improve crude purity [17] [18].
- Washing: Wash the resin with DMF to remove excess reagents.
3. Cleavage and Side Chain Deprotection
- Cleavage Cocktail: Transfer the peptide-bound resin to the precipitating unit. Add a cleavage cocktail such as TFA/Water/TIPS (90:5:5, v/v/v) and mix with low-pressure nitrogen flow for 2 hours [17].
- Alternative for PNA: For PNA conjugates, use TFA containing 5% m-cresol as a scavenger. Cleavage time is dependent on the support (90 min for PAL-PEG-PS, 5 min for XAL handle) [12].
4. Precipitation and Isolation
- Precipitation: In the precipitating unit, add the TFA/peptide mixture to pre-chilled diethyl ether (-20°C) and mix via nitrogen bubbling for 30 min.
- Washing: Filter the precipitate and wash several times with cold diethyl ether.
- Drying: Dry the peptide precipitate under vacuum, then dissolve in MeCN/Hâ‚‚O (1:1, v/v) for analysis [17].
Protocol 2: Forming Non-Covalent CPP-siRNA Complexes for Delivery
This protocol describes the formation of nanocomplexes using amphipathic CPPs like MPG or CADY for siRNA delivery [13].
1. Preparation of Solutions
- CPP Stock: Dissolve the CPP (e.g., CADY) in nuclease-free water or an appropriate buffer at a stock concentration of 100 µM.
- siRNA Stock: Dilute the siRNA to a stock concentration of 20 µM in nuclease-free water.
2. Complex Formation
- Mixing: In a microcentrifuge tube, rapidly mix the CPP and siRNA solutions at a predetermined molar ratio (e.g., 10:1 to 40:1 CPP:siRNA). The optimal ratio should be determined empirically for each CPP-siRNA pair.
- Incubation: Incubate the mixture for 15-30 minutes at 37°C to allow for stable nanoparticle formation.
3. Cellular Assay
- Cell Seeding: Seed cells in a 24-well plate to reach 50-70% confluency at the time of transfection.
- Transfection: Replace the growth medium with fresh medium. Add the CPP-siRNA complexes directly to the cells.
- Incubation: Incubate the cells for 1-4 hours at 37°C, then replace the transfection medium with fresh growth medium.
- Analysis: Assess gene silencing efficiency (e.g., via qRT-PCR or Western Blot) 48-72 hours post-transfection.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CPP and Peptide-PNA Conjugate Research
| Item | Function/Description | Example/Note |
|---|---|---|
| Fmoc-PNA Monomers | Building blocks for automated PNA synthesis on DNA/peptide synthesizers [12]. | Supplied as 0.2M solutions in N-methylpyrrolidone (NMP); may require gentle heating to dissolve [12]. |
| Solid Support (Resin) | A universal support for Fmoc-based synthesis, producing a C-terminal amide [12]. | PAL-PEG-PS or XAL-PEG-PS (low load, <0.2 mmol/g) to avoid aggregation [12]. |
| Coupling Reagents | Activates the carboxylic acid of the monomer for coupling to the growing chain. | HATU or PyBOP, used with a base mixture of DIPEA and lutidine [17] [12]. |
| Cleavage Cocktail | Cleaves the peptide from the resin and removes acid-labile protecting groups. | TFA-based cocktails with scavengers (e.g., TIPS, water, m-cresol, phenol) [17] [12]. |
| Spacer Monomers (e.g., AEEA) | Improves solubility and provides a flexible spacer for labeling [12]. | Incorporation of two or more spacers between the PNA oligomer and a label (e.g., fluorescein, biotin) is recommended [12]. |
| Amphipathic CPPs (MPG, CADY) | For non-covalent delivery of cargoes like siRNA and proteins [13]. | Forms stable, non-covalent complexes that maintain full biological activity of the cargo [13]. |
| AD-8007 | AD-8007, MF:C22H26N2O, MW:334.5 g/mol | Chemical Reagent |
| SPL-IN-1 | SPL-IN-1, MF:C31H42N2O6S2, MW:602.8 g/mol | Chemical Reagent |
Workflow Integration: CPP-PNA Conjugates on an Automated Platform
The synthesis of peptide-PNA conjugates, which combine the gene-targeting ability of PNA with the delivery capability of CPPs, is a prime application for automated synthesizers. These platforms can integrate SPPS with valuable transformations like native chemical ligation (NCL) into a single, uninterrupted protocol [17]. The software's capability to flexibly control reagent movement allows for the automation of novel chemistry, including the synthesis of peptide-nucleic acids [18].
Cell-penetrating peptides are indispensable tools for overcoming the critical barrier of cellular membrane permeability. Their ability to ferry diverse cargoes into cells, combined with advanced automated synthesis platforms, unlocks new possibilities for research and drug development. The protocols and workflows outlined here provide a foundation for the reliable production and application of CPPs and CPP-PNA conjugates, facilitating the advancement of intracellular therapeutics and diagnostics.
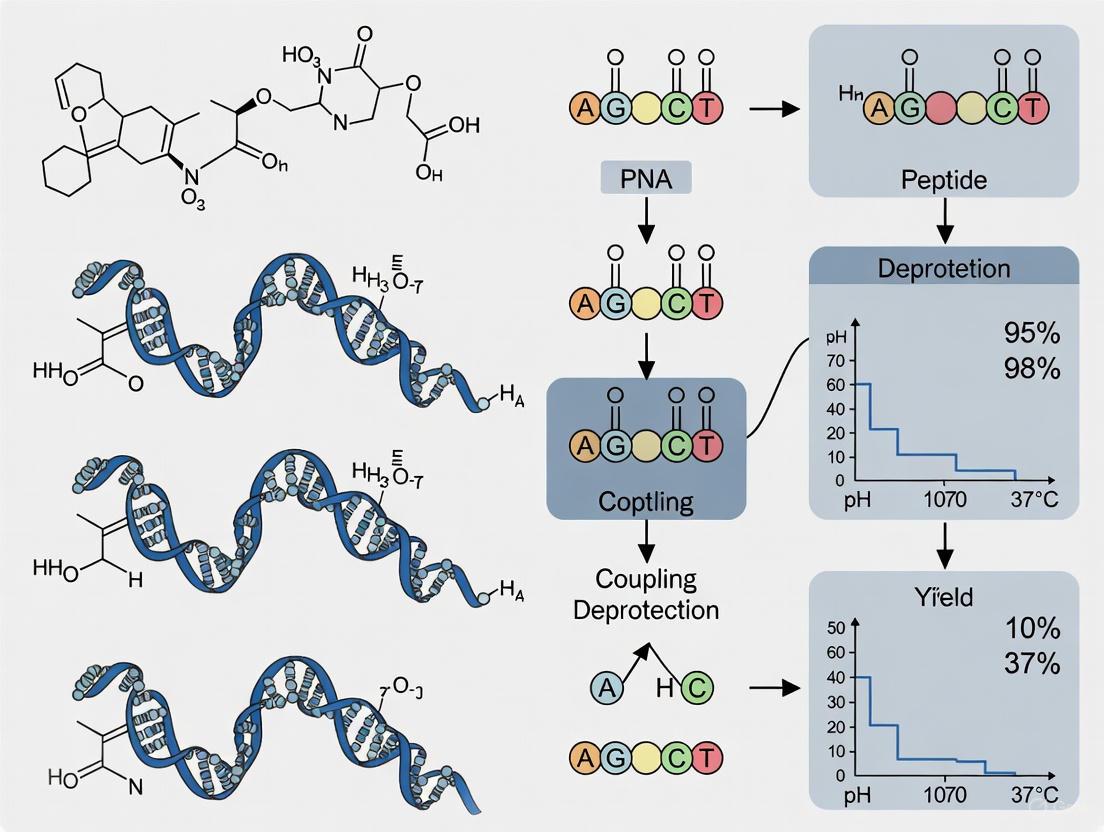
Peptide Nucleic Acids (PNAs) represent a powerful class of synthetic oligonucleotide analogs with exceptional binding affinity and specificity toward complementary DNA and RNA sequences [9]. Their unique N-(2-aminoethyl)glycine backbone replaces the sugar-phosphodiester moiety of natural nucleic acids, conferring superior chemical, thermal, and enzymatic stability [5]. These properties make PNAs particularly valuable for antisense therapeutic development, molecular diagnostics, and biomedical research [9]. However, the transition of PNA-based technologies from research tools to clinical applications has been hampered by significant synthetic challenges inherent to manual and standard batch methods. These limitations constitute a critical bottleneck in PNA research and development, particularly for complex constructs like peptide-PNA conjugates (PPNAs) that require enhanced cellular delivery [5] [9].
The fundamental obstacle in PNA synthesis stems from the inherent properties of the PNA backbone itself. Unlike traditional peptides or oligonucleotides, PNA oligomers exhibit poor solubility and a strong tendency for aggregation, especially for purine-rich sequences [11]. This aggregation problem is exacerbated in standard batch synthesis protocols, where on-resin aggregation leads to decreased coupling efficiency and increased formation of deletion sequences and side products [5]. Additionally, longer PNA sequences (>15-mers) that are often necessary for optimal target affinity and specificity present particular challenges for conventional methods [5]. This application note examines these limitations through quantitative data and presents automated flow synthesis as a transformative solution.
Quantitative Analysis of Synthesis Limitations
Table 1: Comparative Performance of PNA Synthesis Methods
| Synthesis Parameter | Manual Batch Synthesis | Standard Automated Batch | Automated Fast-Flow |
|---|---|---|---|
| Coupling Time per Amide Bond | ~4 hours (for a 4-mer) [5] | 10 minutes (at 45°C) [5] | 10 seconds [5] |
| Crude Purity (4-mer Example) | 57% [5] | Not specified | 70% [5] |
| Typical Maximum Length | <15 monomers [5] [11] | <15 monomers [5] [11] | >15 monomers reliably [5] |
| Key Side Products | ~15% isomers, ~4% deletions, ~2% nucleobase adducts [5] | Higher deletion rates and nucleobase adducts at elevated temperatures [5] | Significantly reduced side products with optimized protocols [5] |
| Compatibility with Conjugate Synthesis | Requires multi-step, off-line conjugation strategies [20] | Limited by on-resin aggregation during extended sequences [5] | Single-shot synthesis of PNA-peptide conjugates [5] |
Table 2: Common Side Reactions in Standard Fmoc-Based PNA Synthesis
| Side Reaction | Cause | Impact on Product Quality | Common Mitigation Strategies |
|---|---|---|---|
| Base-Catalyzed Rearrangement | Extended exposure to piperidine during Fmoc deprotection [11] | ~0.3-0.4% contamination with 2-minute deprotection [11] | Minimize deprotection time; use microwave assistance [11] |
| Aspartimide Formation | Base-mediated side reaction during deprotection [5] | Sequence deletion and isomerization [5] | Addition of formic acid to deprotection solutions [5] |
| Nucleobase Adducts | Particularly problematic during high-temperature synthesis [5] | 7% observed in non-optimized flow synthesis [5] | Optimized deprotection temperature (~40°C) and base [5] |
| On-Resin Aggregation | Inter-chain hydrogen bonding of the PNA backbone [5] | Reduced coupling efficiency, increased deletion sequences [5] | Capping and double-couplings (often insufficient) [5] |
Experimental Protocols for Method Comparison
Protocol A: Manual Solid-Phase PNA Synthesis (Fmoc/Bhoc Chemistry)
This protocol outlines the traditional manual synthesis of a PNA oligomer, highlighting the time-intensive and laborious steps that contribute to the synthesis bottleneck.
- Resin Preparation: Use 100 mg of Rink Amide resin (loading capacity ~0.18 mmol/g). Pre-swell the resin in DMF or NMP for 30 minutes [5] [11].
- Deprotection Cycle: Treat the resin with 20% piperidine in DMF or NMP (2 x 5-10 minutes) to remove the Fmoc protecting group. Monitor deprotection by in-line UV-vis if available [5] [11].
- Coupling Cycle:
- Reagent Preparation: Dissolve 6 equivalents of Fmoc-PNA monomer in NMP. In a separate vessel, prepare a solution of 5.8 equivalents PyAOP activator and 6 equivalents each of DIEA and 2,5-lutidine in NMP [5].
- Reaction: Mix the activated monomer solution with the resin and agitate for 30-60 minutes to complete the coupling reaction [11].
- Capping: Introduce a capping solution (typically acetic anhydride) to block any unreacted chain ends to prevent deletion sequences [5] [11].
- Repetition: Repeat the deprotection and coupling cycles for each monomer in the sequence.
- Cleavage and Global Deprotection: Treat the resin with a cleavage cocktail containing 20% m-cresol in TFA for 2-4 hours to simultaneously cleave the product from the resin and remove Bhoc protecting groups from nucleobases [11].
- Precipitation and Purification: Precipitate the crude PNA in cold diethyl ether, isolate by centrifugation, and purify by reverse-phase HPLC (C18 column) using a water/acetonitrile gradient with 0.1% TFA [11].
Protocol B: Automated Fast-Flow Synthesis of a Peptide-PNA Conjugate
This protocol describes the automated, continuous-flow method that dramatically accelerates the synthesis of PNA and its conjugates.
- Instrument Setup: Utilize a fully automated flow synthesizer (e.g., iChemAFS) equipped with a central control computer, solution storage system, HPLC pumps, multiposition valves, heated reaction zones, and an in-line UV-vis detector [5] [4].
- Reactor Preparation: Pack a reusable reactor with 15 mg of 0.5 mmol/g Rink Amide resin (7.5 μmol scale) [5].
- Automated Synthesis Cycle (3 minutes per monomer):
- Activation and Coupling: Three HPLC pumps simultaneously draw PNA monomer (10 equiv), activator (PyAOP, 9.6 equiv), and base (DIEA, 30 equiv) from storage. The solutions are merged at a valve and passed through a 70°C heating loop to form the activated ester, which then flows through the 70°C resin-packed reactor. Amide bond formation is completed within 10 seconds at a flow rate of 2.5 mL/min [5].
- Deprotection: A piperidine solution is pumped through a room-temperature loop and mixed with the 70°C reactor, creating an optimal ~40°C environment for efficient Fmoc removal while minimizing nucleobase adducts [5].
- Monitoring: The in-line UV-vis detector monitors the Fmoc-piperidine adduct in the spent reagent stream to provide real-time feedback on deprotection efficiency [5].
- Direct Peptide Conjugation: Following PNA chain assembly, switch the reagent reservoirs to Fmoc-amino acids and continue synthesis in the same automated run to covalently attach a cell-penetrating peptide (CPP) sequence, creating the PPNA conjugate in a "single-shot" without intermediate cleavage or purification [5].
- Final Cleavage and Analysis: Cleave the final conjugate from the resin using standard TFA cocktail, precipitate, and characterize by HPLC and LC-MS [5].
Synthesis Method Comparison Diagram
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for PNA Synthesis
| Reagent/Material | Function/Purpose | Application Notes |
|---|---|---|
| Fmoc/Bhoc PNA Monomers | Building blocks for chain assembly; Fmoc protects backbone, Bhoc protects nucleobases [11]. | Most common chemistry for automated synthesizers; compatible with peptide synthesisers [11]. |
| Rink Amide Resin (XAL-PEG-PS) | Solid support for synthesis; provides amide terminus upon cleavage [11] [7]. | Universal support for both PNA and peptide synthesis; PEG spacer improves solvation and yield [11]. |
| PyAOP Activator | Peptide coupling reagent forming active esters for amide bond formation [5]. | Used with DIEA base; preferred for efficient coupling in both manual and flow systems [5]. |
| AEEA Linker (eg1) | Spacer molecule incorporated between PNA and peptide sequences [11]. | Crucial for reducing steric hindrance and improving conjugation efficiency and final yield [11]. |
| Cell-Penetrating Peptides (CPPs) | Peptide vectors (e.g., (KFF)3K, TP10) conjugated to PNA to enable cellular uptake [5] [9] [7]. | Covalently attached to PNA to overcome innate poor membrane permeability [9]. |
| Neostenine | Neostenine, MF:C17H27NO2, MW:277.4 g/mol | Chemical Reagent |
| Tenacissoside G | Tenacissoside G, MF:C42H64O14, MW:792.9 g/mol | Chemical Reagent |
The limitations of manual and standard batch methods for PNA synthesis—characterized by excessively long cycle times, low crude purity, limited access to therapeutically relevant longer sequences, and inefficient conjugate production—represent a significant impediment to the advancement of PNA-based therapeutics and research tools. The data and protocols presented herein quantitatively define this synthesis bottleneck. The adoption of fully automated, high-temperature flow synthesis platforms directly addresses these challenges, reducing coupling times from hours to seconds, enabling the single-shot production of complex PNA-peptide conjugates, and providing researchers with a robust and scalable method to accelerate the discovery and development of next-generation PNA agents.
Inside the Automated Flow Synthesizer: Engineering and Workflow for Rapid PPNA Production
Architecture of a Fully Automated Fast-Flow Instrument
The synthesis of complex biomolecules like peptide-PNA conjugates (PPNAs) presents significant challenges in modern therapeutic development, including low yields, long synthesis times, and sequence limitations. Automated fast-flow synthesis technology has emerged as a transformative approach to overcome these hurdles, enabling rapid production of high-purity compounds for drug discovery applications. This technical note details the architecture and operation of a fully automated fast-flow instrument specifically designed for the synthesis of PPNA conjugates, framing this technology within the broader context of an automated platform for peptide-PNA conjugate research. The platform significantly accelerates the development timeline for antisense therapeutics, particularly relevant for rapid response to emerging viral pathogens such as SARS-CoV-2 [21] [5].
Instrument Architecture and Modules
The automated microscale flow synthesizer is constructed from commercially available components and a machined reaction vessel, employing a design philosophy similar to previously reported fast-flow peptide synthesizers [21]. The instrument architecture comprises six or seven interconnected modules that work in concert to enable fully automated synthesis.
Table 1: Core Modules of the Automated Fast-Flow Synthesizer
| Module Number | Module Name | Key Components | Primary Function |
|---|---|---|---|
| 1 | Reagent Storage | Glass containers, nitrogen atmosphere | Storage and preservation of liquid reagents and PNA monomers |
| 2 | Valve System | Two chemically inert multiposition valves | Selection and direction of reagents from storage |
| 3 | Pump System | Two or three HPLC pumps | Precise delivery and metering of reagent streams |
| 4 | Reaction Zone | Heated metal tube, aluminum core, reactor chamber | Housing solid-phase resin and facilitating coupling reactions |
| 5 | In-line Monitoring | UV-Vis detector | Real-time analysis of spent reagent composition |
| 6 | Control System | Computer, Mechwolf programming environment [21] [5] | Coordination and control of all instrument modules |
The synthesis process begins with reagents stored in Module 1. Module 2 valves select the required reagents, which are then pumped (Module 3) to a T-mixer. The combined flow travels through Module 4, where it is preheated in a metal tube before passing through the resin-packed reactor chamber. The solid-phase resin, typically Rink Amide resin with a loading of 0.5 mmol/g, is contained in a removable reactor chamber with a 1 mL volume, designed for a 4.4-7.5 μmol-scale synthesis [21] [5]. Module 5 monitors the process, and Module 6 orchestrates the entire operation via a modular Python script in the Mechwolf environment [21] [5].
The logical workflow of the synthesis process is controlled by a central script that defines the sequence of operations for each module. The relationship between the command script and the instrument's physical response is critical for achieving high-fidelity synthesis.
Performance Data and Optimization
The transition from traditional manual synthesis to automated fast-flow systems has yielded dramatic improvements in synthesis efficiency. The following table quantifies these performance gains.
Table 2: Performance Comparison of PNA Synthesis Methods
| Synthesis Parameter | Manual Batch Synthesis | Automated Fast-Flow Synthesis |
|---|---|---|
| Coupling Time per Amide Bond | ~10 minutes (45°C microwave) [5] to 180 minutes (PMO, room temp) [21] | 10 seconds [5] to 8 minutes (PMO) [21] |
| Total Synthesis Cycle Time | Several hours for a 4-mer [5] | ~3 minutes per cycle [5], ~3.5 hours for a full therapeutic PMO [21] |
| Typical Crude Purity | ~57% for a 4-mer PNA [5] | Up to 92-95% [21] [4] |
| Synthesis Scale | ~100 mg resin [5] | 4.4-7.5 μmol scale (e.g., 15 mg of 0.5 mmol/g resin) [21] [5] |
| Key Enabler | - | High-temperature (70-90°C) flow chemistry [21] |
Optimization of the synthesis process was achieved through iterative changes in flow variables. A major focus was determining the maximum allowable temperature to accelerate reactions without degrading synthetic intermediates. Initial screens at 70°C provided the desired material but with sub-optimal crude purity of 72%. Further optimization of the instrument command recipe, monomer equivalents, and coupling catalysts improved crude purity to 92% [21]. The use of in-line UV-vis monitoring combined with LC-MS and HPLC product characterization was crucial for this rapid optimization, allowing for direct feedback on deprotection efficiency and mass transfer rates [5].
Experimental Protocol: Fully Automated Flow Synthesis of PPNA
Reagent Preparation and Instrument Setup
Research Reagent Solutions and Essential Materials
Table 3: Key Research Reagents and Materials for PPNA Synthesis
| Reagent/Material | Specification/Function |
|---|---|
| PNA Monomers | Fmoc-protected; building blocks for oligomer synthesis. |
| Activator | e.g., PyAOP ((7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate); facilitates amide bond formation [5]. |
| Base | e.g., DIEA (N,N-Diisopropylethylamine); acts as a proton scavenger [5]. |
| Deprotection Solution | Piperidine in DMF; removes the Fmoc protecting group [5]. |
| Solid Support | Rink Amide Resin (0.5 mmol/g loading); provides the solid phase for chain elongation [5]. |
| Solvent | Anhydrous DMF (Dimethylformamide); primary solvent for reagents. |
- Resin Loading: Load 15 mg of Rink Amide resin (0.5 mmol/g loading) into the removable reactor chamber. This establishes a 7.5 μmol scale synthesis [5].
- Reagent Preparation: Prepare and load all reagent solutions into the designated glass containers in Module 1 under a nitrogen atmosphere:
- System Initialization: Power on the HPLC pumps (Module 3), heating elements for the reaction zone (Module 4), and the UV-vis detector (Module 5). Initialize the control computer (Module 6) and load the synthesis script.
Synthesis Execution and Cyclic Operation
The following workflow details a single synthesis cycle for the addition of one PNA monomer. This cycle is repeated iteratively for each base in the target sequence.
Fmoc Deprotection
- Action: The control script activates the valve (Module 2) to select the deprotection solution (piperidine in DMF). The HPLC pump (Module 3) delivers the solution through the reaction zone (Module 4). For high-temperature synthesis, the piperidine solution may flow through a room-temperature loop before meeting the preheated reactor to create an optimal ~40°C deprotection environment [5].
- Purpose: Removes the Fmoc (fluorenylmethyloxycarbonyl) protecting group from the terminal amine of the growing PNA chain, enabling the next coupling reaction.
- Monitoring: The UV-vis detector (Module 5) monitors the characteristic absorbance of the removed Fmoc group, providing data on deprotection efficiency [5].
Washing
- Action: The system switches to deliver a wash solvent (e.g., DMF) to flush the reactor and resin bed clean of the deprotection solution and by-products. A wash volume of at least 1.6 mL (20 pump strokes) is typically used to ensure complete clearance [21].
Monomer Coupling
- Action: The valve system (Module 2) simultaneously selects the streams for the PNA monomer, activator (e.g., PyAOP), and base (e.g., DIEA). The three HPLC pumps (Module 3) deliver these reagents, which are merged at a T-mixer. The mixture flows through a preheater, forming an activated ester, and then through the resin-packed reactor chamber maintained at high temperature (70-90°C) [21] [5].
- Critical Parameters: The coupling reaction is completed in as little as 10 seconds per amide bond at a flow rate of 2.5 mL/min [5]. The high temperature is a key factor in achieving this dramatic rate enhancement.
Washing
- Action: The system performs another wash cycle with an appropriate solvent to remove excess monomer and coupling reagents from the resin bed.
Cycle Repetition and Completion
- Steps 1-4 constitute one coupling cycle and are repeated automatically for each monomer in the target PNA sequence.
- Peptide Conjugation (for PPNAs): Following PNA chain assembly, a cell-penetrating peptide (CPP) can be synthesized directly on the PNA chain in a "single-shot" process using the same instrument and coupling protocol, without intermediate cleavage [5].
- Final Cleavage: Once the full sequence is assembled, the flow of a cleavage cocktail (e.g., Trifluoroacetic acid-based solutions) through the reactor severs the completed PPNA from the solid support and removes any remaining protecting groups, yielding the crude product in solution.
Post-Synthesis Analysis
- Product Analysis: Analyze the crude product using Liquid Chromatography-Mass Spectrometry (LC-MS) and High-Performance Liquid Chromatography (HPLC) to determine purity and identity. The optimized flow synthesis protocol achieves crude purities of up to 92-95% for target sequences [21] [4].
- Bioactivity Testing: For therapeutic candidates, perform relevant biological assays. For example, anti-SARS-CoV-2 PPNAs synthesized with this technology have been evaluated in live virus infection assays, demonstrating over 95% reduction in viral titer [5].
The fully automated fast-flow synthesizer represents a significant advancement in the architecture for biomolecule synthesis. Its modular design, integration of high-temperature flow chemistry, and comprehensive computer control enable the rapid and efficient production of challenging molecules like peptide-PNA conjugates. This platform reduces synthesis times from days to hours, provides high crude purity, and facilitates the direct "single-shot" synthesis of complex conjugates. By automating and accelerating the construction of these potential therapeutics, this technology serves as a powerful tool in an automated peptide-PNA research platform, significantly accelerating the drug discovery pipeline for applications from genetic diseases to antiviral agents.
Within the field of peptide-peptide nucleic acid (PNA) conjugate research, the synthesis of these complex molecules has been a significant bottleneck. Traditional solid-phase synthesis methods are time-consuming, often requiring 60 to 100 minutes to incorporate a single amino acid residue [22]. This technical limitation has severely restricted the rapid investigation and development of PNA-based therapeutics, including antisense agents for applications such as antiviral and anticancer therapies [5]. However, recent advances in automated flow chemistry have revolutionized this landscape, enabling the formation of amide bonds in as little as 10 seconds under optimized high-temperature conditions [5]. This application note details the protocols and quantitative data supporting this transformative methodology, providing researchers with the tools to implement accelerated synthesis within their automated platforms for peptide-PNA conjugate production.
Key Principles and Quantitative Performance Data
The foundation of ultra-rapid amide bond formation lies in the precise application of elevated temperature and continuous flow dynamics. At room temperature, amide bond formation using activated amino acid solutions is already rapid, with a reaction half-life of approximately 4.6 seconds [22]. The application of high-temperature flow chemistry intensifies this process dramatically; at 60°C, amide bond formation is complete in less than 10 seconds, and Fmoc removal occurs in less than 20 seconds [22]. Further optimization has demonstrated that a temperature of 70°C is optimal for PNA synthesis specifically, enabling each amide bond to be formed in precisely 10 seconds [5]. This represents an order-of-magnitude acceleration compared to commercial microwave synthesizers (10 minutes/amide bond) or traditional DNA synthesizers (32 minutes/coupling cycle) [5].
Table 1: Comparative Performance of Amide Bond Formation Methodologies
| Synthesis Method | Temperature | Time per Amide Bond | Relative Speed |
|---|---|---|---|
| Traditional Fmoc SPPS [22] | Room Temperature | 60-100 minutes | 1x |
| Microwave-Assisted SPPS [5] | 45°C | 20 minutes | 3-5x |
| Early Flow-Based SPPS [22] | 60°C | 12 minutes | 5-8x |
| High-Temperature Flow Chemistry (PNA) [5] | 70°C | 10 seconds | 360-600x |
The accelerated kinetics are not solely temperature-dependent. Flow-based systems maintain a maximal concentration of reagents in the reaction vessel, quickly exchange reagents, and eliminate the time needed to heat reagents after they have been added to the vessel [22]. This integrated approach of high temperature and optimized fluidics enables the complete solid-phase PNA synthesis cycle, including washing and deprotection steps, to be accomplished within 3 minutes per monomer [5].
Table 2: Impact of High-Temperature Flow Synthesis on Complete PNA Production
| Metric | Traditional Synthesis | High-Temperature Flow | Improvement |
|---|---|---|---|
| Cycle Time per Residue | ~90 minutes [22] | ~3 minutes [5] | 30x faster |
| Synthesis Time for 18-mer PPNA | ~27 hours | ~54 minutes | 30x faster |
| Daily Output (Theoretical) | <1 complete PPNAs | 8 anti-SARS-CoV-2 PPNAs [5] | 8x higher throughput |
| Crude Purity (Exemplary 4-mer) | 57% [5] | 70% [5] | Significant enhancement |
Experimental Protocols
Automated Flow Synthesis Apparatus Configuration
The automated flow platform for high-speed PNA synthesis consists of an integrated system with specific components that work in concert to achieve the rapid amide bond formation.
Step-by-Step Synthesis Protocol
Protocol: Automated High-Temperature Flow Synthesis of PNA-Peptide Conjugates
Materials and Equipment:
- Automated flow synthesizer (e.g., iChemAFS platform or equivalent) [4] [5]
- Rink Amide resin (0.5 mmol/g loading) [5]
- Fmoc/Bhoc protected PNA monomers
- Activator: PyAOP or HATU/HBTU
- Base: DIEA (N,N-Diisopropylethylamine)
- Deprotection reagent: 50% (v/v) piperidine in DMF
- Anhydrous DMF solvent
Procedure:
Reactor Setup
- Load 15 mg of Rink Amide resin (0.5 mmol/g) into the custom reactor designed for 7.5 μmol scale synthesis [5].
- Ensure the reactor is properly packed to create a packed bed with low backpressure.
System Pre-conditioning
- Pre-heat the entire system, including the heat exchanger and reactor, to the target temperature of 70°C [5].
- Purge all fluidic pathways with anhydrous DMF to eliminate moisture and ensure solvent compatibility.
Coupling Cycle (10 seconds per amide bond)
- Prepare the coupling solution containing 10 equivalents of PNA monomer, 9.6 equivalents of activator (PyAOP), and 30 equivalents of DIEA base in anhydrous DMF [5].
- Deliver the coupling solution through the pre-heat loop (70°C) at a flow rate of 2.5 mL/min.
- Pass the activated monomer through the heated reactor (70°C) for precisely 10 seconds to complete amide bond formation [5].
- Monitor the process using in-line UV-vis detection to verify reagent delivery.
Washing Step
- Wash the resin with 20 mL of DMF delivered over 2 minutes at a flow rate of 10 mL/min [22].
- Ensure complete removal of excess coupling reagents before proceeding to deprotection.
Fmoc Deprotection
- Deliver 50% (v/v) piperidine in DMF through a room temperature loop, which then meets the 70°C reactor to create an approximate 40°C environment [5].
- Maintain deprotection conditions for the optimized time (determined empirically, typically 30-60 seconds).
- Monitor the deprotection efficiency via UV absorbance at 304 nm, tracking the formation of the dibenzofulvene-piperidine adduct [22] [5].
Repeat Cycle
- Repeat steps 3-5 for each additional monomer in the sequence.
- For peptide-PNA conjugates (PPNAs), continue synthesis seamlessly by switching to Fmoc-protected amino acids after completing the PNA sequence [5].
Cleavage and Purification
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of high-temperature flow synthesis for PNA conjugates requires specific reagents and materials optimized for these accelerated conditions.
Table 3: Essential Research Reagent Solutions for High-Temperature PNA Flow Synthesis
| Reagent/Material | Function | Optimized Concentration/Type | Notes |
|---|---|---|---|
| PNA Monomers | Building blocks for PNA synthesis | Fmoc/Bhoc protected [7] | Bhoc protection of nucleobases prevents side reactions during synthesis |
| Activator | Activates carboxylic acid for amide coupling | PyAOP, HATU, or HBTU [5] | 9.6 equivalents relative to monomer; PyAOP particularly effective for PNA |
| Base | Facilitates activation and coupling | DIEA (N,N-Diisopropylethylamine) [5] | 30 equivalents in DMF; critical for maintaining coupling efficiency |
| Deprotection Reagent | Removes Fmoc protecting group | 50% (v/v) piperidine in DMF [22] [5] | Higher concentration than standard 20% for faster deprotection at elevated temperature |
| Solid Support | Platform for solid-phase synthesis | Rink Amide resin (0.5 mmol/g) [5] | Higher loading resin (0.5 mmol/g) enables microscale synthesis (7.5 μmol) |
| Coupling Solvent | Reaction medium | Anhydrous DMF [5] | Must be rigorously anhydrous to prevent premature hydrolysis of activated esters |
| DAN Linker | Enables on-resin cyclization | Diaminonicotinic acid (DAN) [4] [6] | Critical for head-to-tail cyclization of cyclic peptide-PNA conjugates |
| AT1R antagonist 3 | AT1R antagonist 3, MF:C22H16N6O2S, MW:428.5 g/mol | Chemical Reagent | Bench Chemicals |
| Coenzyme FO | Coenzyme FO, MF:C16H17N3O7, MW:363.32 g/mol | Chemical Reagent | Bench Chemicals |
Applications and Case Studies
The implementation of this high-temperature flow synthesis platform has enabled significant advances in PNA research and therapeutic development. A primary demonstration involved the synthesis of an 18-mer PPNA targeting the β-thalassemia gene sequence (IVS2-654) conjugated to a 12-mer cell-penetrating peptide in a single, automated operation [5]. The resulting conjugate exhibited threefold higher activity in splice-correction assays compared to transfected PNA, demonstrating that the rapid synthesis method produces biologically active molecules [5].
Perhaps the most compelling validation of the platform's throughput came during the SARS-CoV-2 pandemic, when researchers synthesized eight different anti-SARS-CoV-2 PPNAs in a single day [5]. One of these conjugates, targeting the 5' untranslated region of the SARS-CoV-2 genomic RNA, reduced viral titer by over 95% in live virus infection assays (IC₅₀ = 0.8 μM) [5] [23]. This case study highlights how the accelerated synthesis capability enables rapid response to emerging health threats by facilitating the quick production and screening of multiple therapeutic candidates.
Beyond linear conjugates, the methodology has been extended to cyclic peptide-PNA conjugates using an automated on-resin head-to-tail cyclization strategy with a diaminonicotinic acid (DAN) linker [4] [6]. This approach has achieved 2- to 12-residue cyclic sequences with up to 95% crude purity and has been used to construct libraries of 22 PNA/PPNA structures for membrane permeability screening [4]. The technology has also been applied to synthesize cyclic PPNA nanotubes with high aspect ratios, characterized by TEM and AFM, demonstrating applications in nanotechnology as well as biomedicine [4] [6].
The development of antisense oligonucleotide-based therapeutics represents a rapidly advancing frontier in biomedical science, particularly for treating genetic, viral, and acquired diseases [5]. Among these emerging biologics, peptide nucleic acids (PNAs) offer unique advantages due to their charge-neutral pseudopeptide backbone, which confers superior binding affinity and enzymatic stability compared to natural nucleic acids [5]. However, the clinical translation of PNAs has been hampered by challenges including poor cellular uptake, limited solubility, and rapid elimination [5]. While conjugation to cell-penetrating peptides (CPPs) can mitigate these limitations, the synthesis of these complex conjugates—particularly longer PNA sequences (>15-mer)—remains inefficient using conventional methods [5].
Traditional batch synthesis approaches often struggle with on-resin aggregation, deletion sequences, and various side reactions, making robust production of long PNA sequences difficult [5]. The established workaround of stepwise synthesis via click chemistry adds complexity and reduces overall efficiency [5]. This application note details a transformative fully automated fast-flow synthesis platform that enables single-shot manufacturing of CPP-PNA conjugates (PPNAs) with dramatically improved speed and purity, effectively overcoming previous synthetic limitations and accelerating therapeutic development.
Automated Fast-Flow Synthesis Platform
Instrument Design and Workflow
The automated flow PNA synthesizer represents a significant engineering advancement, comprising seven integrated modules: a central control computer, solution storage system, three HPLC pumps, three multiposition valves, heating elements, reaction zone, and an in-line UV–vis detector [5]. The system operates under the control of a modular script in the Mechwolf programming environment, coordinating all synthetic processes with precision [5].
During the coupling reaction, three HPLC pumps simultaneously draw reagents (PNA monomer, activator, and base solutions) from nitrogen-purged storage reservoirs [5]. These solutions merge via a valve assembly and pass through a 70°C heating module, where the PNA monomer rapidly forms an activated ester. This activated species then flows through the reaction zone—a packed bed of solid-phase resin maintained at 70°C—where amide bond formation completes within merely 10 seconds [5]. The deprotection step utilizes a temperature-gradient approach, with piperidine solution flowing through a room temperature loop before meeting the 70°C reactor to create an optimal ~40°C deprotection environment that minimizes nucleobase adduct formation [5].
The synthesizer employs a reusable reactor body designed for 7.5 μmol scale synthesis, typically loaded with 15 mg of 0.5 mmol/g Rink Amide resin [5]. This microscale approach significantly reduces expensive monomer consumption while yielding sufficient product (milligrams) for comprehensive biological characterization. The complete synthetic cycle for each amide bond formation requires approximately 3 minutes, representing a substantial improvement over conventional methods [5].
The following workflow diagram illustrates the automated synthesis process:
Synthesis Optimization and Efficiency
Extensive optimization efforts have established the superior performance of the automated flow synthesis platform compared to manual methods. The evaluation of reaction conditions revealed significant improvements in crude purity and reduction of side products [5].
Table 1: Optimization of Automated Flow PNA Synthesis Conditions
| Entry | Synthesis Method | Temperature | Deprotection Base | Crude Purity | Major Side Products |
|---|---|---|---|---|---|
| 1 | Manual | ~25°C | Piperidine | 57% | ~15% isomers, ~4% deletions, ~2% nucleobase adducts |
| 2 | Automated Flow | 70°C | Piperidine | 70% | ~10% deletions, ~7% nucleobase adducts |
| 3 | Automated Flow | 70°C | Piperazine | <60% | Increased deletion sequences |
| 4 | Automated Flow | 70°C | Morpholine | <60% | Increased deletion sequences |
Note: PNA sequence ACTG-Gly-CONHâ‚‚ used for optimization study. Automated conditions: 15 mg Rink Amide resin (0.5 mmol/g), 10 equiv PNA monomer, 9.6 equiv activator, 30 equiv DIEA in DMF, flow rate: 2.5 mL/min. Crude purity determined by HPLC UV absorbance at 280 nm [5].
The high-temperature (70°C) flow conditions significantly accelerate amide bond formation while reducing on-resin aggregation—a major limitation in traditional PNA synthesis [5]. Through systematic screening of deprotection bases, piperidine was identified as optimal for Fmoc-removal at elevated temperatures [5]. A key focus during optimization was preventing piperidine adduct formation during deprotection steps, which was achieved through precise temperature control in the reaction zone [5].
Research Reagent Solutions
The successful implementation of single-shot PPNA synthesis requires specific high-quality reagents and materials. The following table details essential components for establishing this automated platform in a research setting.
Table 2: Essential Research Reagents for Automated PPNA Synthesis
| Reagent/Material | Function/Application | Specifications/Notes |
|---|---|---|
| Rink Amide Resin | Solid support for synthesis | 0.5 mmol/g loading capacity; 15 mg per 7.5 μmol synthesis [5] |
| PNA Monomers | Building blocks for chain elongation | N-(2-aminoethyl)glycine backbone with nucleobase attachments; 10 equivalents per coupling [5] |
| PyAOP Activator | Amide bond formation | (7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate; 9.6 equivalents per coupling [5] |
| DIEA Base | Coupling reaction base | N,N-Diisopropylethylamine; 30 equivalents in DMF [5] |
| Piperidine Solution | Fmoc-deprotection | 20-30% in DMF; optimized for high-temperature deprotection [5] |
| DMF Solvent | Primary reaction solvent | Anhydrous, amine-free quality for optimal coupling efficiency [5] |
| Cell-Penetrating Peptides | Conjugation for enhanced delivery | Covalently attached to PNA; improves cellular uptake and solubility [5] |
Experimental Protocols
Single-Shot PPNA Synthesis Protocol
Objective: To synthesize CPP-conjugated PNAs (PPNAs) in a single, fully automated process using fast-flow technology.
Materials Preparation:
- Resin Preparation: Load 15 mg of Rink Amide resin (0.5 mmol/g capacity) into the reusable reactor body [5].
- Reagent Preparation: Transfer the following solutions to nitrogen-purged reservoirs:
- PNA monomer solutions: 100 mM in DMF (10 equivalents per coupling)
- Activator solution: 96 mM PyAOP in DMF (9.6 equivalents per coupling)
- Base solution: 300 mM DIEA in DMF (30 equivalents per coupling)
- Deprotection solution: 30% (v/v) piperidine in DMF [5]
- System Priming: Prime all fluidic pathways with appropriate solvents, ensuring bubble-free operation.
Synthetic Procedure:
- Initialization: Initiate the Mechwolf control script to begin the automated synthesis sequence [5].
- Deprotection Cycle: Flow piperidine solution through the resin bed at ~40°C for efficient Fmoc removal (monitored via in-line UV-vis at 280 nm) [5].
- Washing: Flush the system with DMF to remove deprotection byproducts completely.
- Coupling Cycle: Simultaneously pump PNA monomer, activator, and base solutions through the 70°C heating module, then through the 70°C reactor for amide bond formation (10 seconds per coupling) [5].
- Repetition: Repeat steps 2-4 for each monomer addition in the target sequence.
- CPP Conjugation: Following PNA chain assembly, proceed with CPP coupling using the same optimized conditions without intermediate cleavage.
- Cleavage and Deprotection: Upon sequence completion, cleave the finished PPNA from the resin using standard TFA-based cocktails.
- Purification: Purify crude products via reversed-phase HPLC and verify by LC-MS analysis.
Critical Parameters:
- Maintain precise temperature control (70°C for coupling, ~40°C for deprotection)
- Ensure complete dissolution of all reagents in high-purity DMF
- Monitor UV chromatograms for consistent Fmoc-removal efficiency throughout synthesis
- Maintain nitrogen atmosphere over reagent solutions to prevent degradation
PPNA Solubilization and Handling
Following synthesis, proper handling of PPNA products is essential for maintaining stability and functionality:
- Reconstitution: Dissolve purified PPNA oligomers in pure deionized water. For difficult sequences, heat the aqueous solution to 60°C for 10 minutes to improve solubility [24].
- Alternative Solvents: If water alone proves insufficient, add 0.1% TFA or 10-20% acetonitrile to the aqueous solution. For particularly challenging cases, DMF or NMP may be used, though these should be avoided if the PNA is intended for PCR applications [24].
- Storage: Prepare stock solutions at 0.1-1 mM concentrations in appropriate solvents, aliquot, and store at -20°C for long-term preservation.
Applications and Biological Validation
The utility of single-shot synthesized PPNAs has been demonstrated across multiple biological applications, establishing this platform as a robust method for producing therapeutically relevant oligonucleotides.
Splice-Correction Application
A synthesized 18-mer PPNA targeting the β-thalassemia IVS2-654 gene sequence displayed threefold enhanced activity compared to transfected unmodified PNA in a splice-correction assay using an enhanced green fluorescence protein (EGFP) reporter system [5]. This demonstrates the significant biological advantage conferred by the CPP conjugation and optimized synthesis.
Experimental Protocol for Splice-Correction Assay:
- Cell Culture: Maintain HeLa pLuc705 cells (which harbor an aberrantly spliced luciferase gene) under standard conditions.
- Transfection: Seed cells in 24-well plates at appropriate density and incubate for 24 hours.
- PPNA Treatment: Apply single-shot synthesized PPNA (1-10 μM range) using commercial transfection reagents or via direct uptake.
- Incubation: Continue culture for 24-48 hours to allow splice correction.
- Analysis: Harvest cells and measure luciferase activity using standard luminescence assays. Normalize data to protein content or control transfection.
Antiviral Application Against SARS-CoV-2
The platform's capability for rapid therapeutic development was demonstrated through the synthesis of eight anti-SARS-CoV-2 PPNAs within a single day [5]. From this library, a PPNA targeting the 5' untranslated region (5'UTR) of SARS-CoV-2 genomic RNA achieved over 95% reduction in viral titer (IC₅₀ = 0.8 μM) in live virus infection assays [5].
Experimental Protocol for Antiviral Assessment:
- Virus and Cells: Culture Vero E6 cells and maintain SARS-CoV-2 virus stocks under appropriate biosafety containment.
- Infection Model: Pre-treat cells with PPNA (0.1-10 μM range) for 4-6 hours before infection with SARS-CoV-2 at low MOI.
- Post-Infection: Maintain PPNA in medium throughout the infection period.
- Viral Titer Quantification: At 48 hours post-infection, collect supernatants and quantify viral titers by plaque assay or TCIDâ‚…â‚€.
- Cytotoxicity Assessment: Perform parallel MTT or similar assays to confirm PPNA treatments do not affect cell viability at active concentrations.
The following diagram illustrates the therapeutic mechanism of antisense PPNAs against viral targets:
Comparative Performance Data
The automated flow synthesis platform demonstrates remarkable efficiency gains compared to conventional methods, as quantified through direct comparison of synthesis metrics and biological outcomes.
Table 3: Performance Comparison of PNA Synthesis Technologies
| Parameter | Manual Batch Synthesis | Commercial Microwave Synthesizer | Automated Fast-Flow Platform |
|---|---|---|---|
| Coupling Time per Amide Bond | ~4 hours for 4-mer [5] | 10 minutes [5] | 10 seconds [5] |
| Typical Crude Purity (15-mer) | ~57% (4-mer) [5] | Not specified | >70% [5] |
| Long Sequence Capability | Challenging (>15-mer) [5] | Limited | Enabled (>15-mer) [5] |
| CPP Conjugation Approach | Stepwise via click chemistry [5] | Stepwise | Single-shot direct synthesis [5] |
| Library Production Time | Days-Weeks | Days | 8 PPNAs in 1 day [5] |
| Biological Activity | Baseline | Not specified | 3-fold enhancement over transfected PNA [5] |
Platform Extensions: Cyclic PNA Synthesis
The automated flow synthesis approach has been successfully extended to cyclic PNA and PPNA structures, which offer enhanced nuclease resistance and binding affinity [4]. Using an automated on-resin head-to-tail cyclization strategy with a diaminonicotinic acid (DAN) linker, researchers have achieved 2- to 12-residue cyclic sequences with up to 95% crude purity [4]. This methodology supports extensive functionalization with natural and non-natural amino acids, enabling construction of diverse libraries for drug discovery and nanotechnology applications [4].
A library of 22 cyclic PNA/PPNA structures has been successfully synthesized and screened for membrane permeability using parallel artificial membrane permeability assays (PAMPA), demonstrating the platform's utility in structure-activity relationship studies [4]. Additionally, the synthesis of cyclic PPNA nanotubes with high aspect ratios highlights the material science applications enabled by this versatile technology [4].
Peptide Nucleic Acids (PNAs) are synthetic oligonucleotide analogues featuring a neutral N-(2-aminoethyl)glycine backbone that confers superior binding affinity, nuclease resistance, and enzymatic stability compared to natural nucleic acids [5] [25] [7]. The conjugation of PNAs with various functional moieties, particularly cell-penetrating peptides (CPPs), creates multifunctional hybrids that overcome inherent limitations of native PNAs, including poor cellular uptake and limited aqueous solubility [5] [26] [7]. Automated synthesis platforms, especially those utilizing fast-flow technology, have revolutionized the production of these conjugates, enabling rapid exploration of their applications across antiviral therapy, antimicrobial interventions, and advanced nanomaterials [5] [27]. This document provides detailed application notes and experimental protocols for leveraging peptide-PNA conjugates (PPNAs) in these diverse research domains.
The following tables summarize key quantitative data demonstrating the efficacy of PNA conjugates across various applications, providing researchers with benchmark performance metrics.
Table 1: Antiviral Efficacy of Selected PNA Conjugates
| Target Virus | PNA Conjugate Type | Target Genomic Region | Efficacy (Viral Titer Reduction/ICâ‚…â‚€) | Citation |
|---|---|---|---|---|
| SARS-CoV-2 | CPP-PPNA | 5' Untranslated Region (5'UTR) | >95% reduction (IC₅₀ = 0.8 μM) | [5] |
| SARS-CoV-2 | CPP-PPNA Library | Various genomic regions | 8 conjugates synthesized and screened in 1 day | [5] [28] |
| Experimental Model | Cell Line / Assay | Primary Readout | Additional Notes | |
| Live virus infection assay | Vero E6 cells | Viral titer quantification | Conjugates synthesized via automated fast-flow | [5] |
Table 2: Design and Performance of PNA Conjugates in Therapeutics and Diagnostics
| Application Domain | PNA Conjugate Design | Biological Target | Key Performance Outcome | Citation |
|---|---|---|---|---|
| Gene Silencing | CPP-PPNA (TP10, Tat, TD2.2) | ACLY, PCSK9 genes | Effective nuclear delivery; TP10 most effective CPP | [7] |
| Splice Correction | Anti-IVS2-654 CPP-PPNA | β-thalassemia IVS2-654 gene | 3-fold activity vs. transfected PNA (EGFP assay) | [5] |
| Cancer Diagnostics | cpG+ modified FIT-PNA | lncRNA CCAT-1 | 2-fold brightness increase (BR=16.9) for RNA detection | [3] |
| Nanomaterial Synthesis | Cyclic PPNA nanotubes | Self-assembly | High aspect ratio structures confirmed by TEM/AFM | [27] |
Experimental Protocols
Protocol 1: Automated Fast-Flow Synthesis of CPP-PNA Conjugates (PPNAs)
This protocol describes the fully automated synthesis of cell-penetrating peptide-conjugated PNAs using a fast-flow instrument, achieving amide bond formation in 10 seconds and completing an 18-mer bioactive PNA within approximately one hour [5] [28].
Research Reagent Solutions
Table 3: Essential Reagents for Automated Flow PNA Synthesis
| Reagent / Material | Function / Role | Specifications / Notes |
|---|---|---|
| Rink Amide Resin | Solid synthesis support | 0.5 mmol/g loading capacity; 15 mg per 7.5 μmol scale synthesis [5] |
| Fmoc-PNA Monomers | Building blocks for chain elongation | Used with Bhoc-protected nucleobases [5] [3] |
| PyAOP Activator | Coupling reagent | (7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate [5] |
| DIEA Base | Activator and coupling base | N,N-Diisopropylethylamine in DMF [5] |
| Piperidine Solution | Fmoc deprotection | 20% in DMF; efficient removal of Fmoc protecting groups [5] |
| DMF Solvent | Primary reaction solvent | Dry, purified, and stored under nitrogen atmosphere [5] |
Step-by-Step Procedure
- Instrument Setup: Initialize the automated flow synthesizer (e.g., iChemAFS or Mechwolf-based system). Ensure three HPLC pumps, multiposition valves, heating elements, and the packed bed reactor are operational. Program the control computer with the modular script for the desired PNA sequence [5] [27].
- Reactor Preparation: Pack the reusable reactor body with 15 mg of Rink Amide resin (0.5 mmol/g). Secure the reactor in the instrument's reaction zone maintained at 70°C for coupling steps [5].
- Coupling Cycle:
- Pumps draw reagents from nitrogen-purged reservoirs: PNA monomer (10 equiv), PyAOP activator (9.6 equiv), and DIEA (30 equiv) in DMF.
- Solutions merge via valve and flow through a 70°C heating loop, forming the activated ester.
- The activated mixture passes through the heated resin bed (70°C), completing amide bond formation within 10 seconds at a flow rate of 2.5 mL/min [5].
- Deprotection Cycle: Piperidine solution flows through a room-temperature loop, mixing with the 70°C reactor environment to achieve ~40°C for efficient Fmoc removal while minimizing nucleobase adduct formation [5].
- In-line Monitoring: Utilize the UV-vis detector to monitor Fmoc-removal absorbance chromatograms, inferring deprotection efficiency and mass transfer rates [5].
- Cleavage and Purification: After sequence completion, cleave the PPNA from the resin using a TFA-based cocktail (e.g., 90:10 TFA/m-cresol for 2 hours). Precipitate the crude product in cold ether, then purify by reverse-phase HPLC (C18 column) with a water/acetonitrile gradient containing 0.1% TFA. Verify identity by LC-MS or ESI-MS [5] [3].
Automated Fast-Flow Synthesis of PPNAs
Protocol 2: Ferritin Nanocage Encapsulation of PNAs for Targeted Delivery
This protocol outlines a divalent-cation-triggered oligomerization method for loading negatively charged PNAs into a polycationic humanized archaeoglobus ferritin (HumAfFt) system, enabling efficient cellular uptake via transferrin receptor 1 (TfR1) mediated endocytosis [25].
Research Reagent Solutions
Table 4: Essential Reagents for PNA Ferritin Encapsulation
| Reagent / Material | Function / Role | Specifications / Notes |
|---|---|---|
| PA3.2-HumAfFt | Polycationic protein cage | HumAfFt chemically modified with cyclic polyamine PA3.2 on interior cysteines [25] |
| PNA Oligomers | Cargo for encapsulation | Can be synthesized with varying lengths and net charges (positive to negative) [25] |
| MgClâ‚‚ or CaClâ‚‚ | Trigger for cage assembly | Divalent cations induce oligomerization of ferritin subunits at physiological pH [25] |
| Dialysis Membrane | Buffer exchange | Standard molecular weight cutoff suitable for protein (e.g., 50 kDa) [25] |
| FITC-labeled PNA | Tracking cargo loading and uptake | Optional; for monitoring encapsulation efficiency and cellular delivery [25] |
Step-by-Step Procedure
- PNA Preparation: Synthesize and purify PNA sequences (e.g., 10-mer or 19-mer) with desired charge characteristics using standard Fmoc solid-phase protocols [25]. Determine concentration spectrophotometrically using calculated molar extinction coefficients.
- Ferritin Modification: Chemically modify HumAfFt with the PA3.2 polycationic linker via thiol-selective reaction using pentafluorobenzene-based derivatives to create a positively charged interior cavity [25].
- Disassembly: Dialyze PA3.2-HumAfFt against a chelating buffer (e.g., 0.5 M EDTA, pH ~8.0) to dissociate the 24-mer ferritin cage into subunits.
- Encapsulation: Mix the ferritin subunits with the PNA cargo at the desired molar ratio in the presence of 150 mM NaCl. Initiate cage reassembly by adding MgClâ‚‚ or CaClâ‚‚ to a final concentration of 10-20 mM. Incubate for 1-2 hours at room temperature [25].
- Purification: Remove unencapsulated PNA and excess salts by dialysis against a physiological buffer (e.g., PBS) or using size exclusion chromatography.
- Characterization: Analyze the PNA-loaded ferritin nanocages using native PAGE, dynamic light scattering (DLS) for size, and HPLC or fluorescence measurement to determine loading efficiency [25].
- Cellular Uptake: Apply purified PNA-loaded ferritin to target cells (e.g., cancer cells overexpressing TfR1). Monitor uptake via confocal microscopy (for FITC-labeled PNA) or evaluate biological activity (e.g., GAPDH gene silencing efficiency) [25].
PNA Encapsulation in Ferritin Nanocages
Protocol 3: Evaluation of PNA Conjugates in Antiviral Assays
This protocol describes the evaluation of synthesized PPNAs against viral targets, specifically using SARS-CoV-2 as a model system, measuring viral titer reduction in a live virus infection assay [5].
Research Reagent Solutions
- PPNA Library: Synthesize PPNAs targeting key viral genomic regions (e.g., 5'UTR, leader sequence, frameshift stimulation element) using Protocol 1 [5] [28].
- Cell Culture: Vero E6 cells (or other susceptible cell lines) maintained in appropriate medium (e.g., DMEM with 10% FBS) [5].
- Virus Stock: SARS-CoV-2 virus stock, handled in BSL-3 containment.
- Plaque Assay Reagents: Overlay medium (e.g., MEM with Avicel), crystal violet staining solution.
Step-by-Step Procedure
- Cell Seeding: Seed Vero E6 cells in 24-well or 48-well plates to reach 90-95% confluency at time of infection.
- PPNA Treatment: Dilute PPNAs to desired concentrations (e.g., 0.1-10 μM range) in serum-free medium. Pre-treat cells with PPNA solutions for 1-4 hours before infection. Include untreated and scramble-PPNA controls.
- Virus Infection: Incubate cells with SARS-CoV-2 at a low multiplicity of infection (MOI, e.g., 0.01) for 1 hour. Remove viral inoculum and replace with fresh maintenance medium containing the respective PPNAs.
- Incubation: Incubate infected cells for 24-48 hours post-infection.
- Viral Titer Quantification:
- Plaque Assay: Collect culture supernatants. Serially dilute and inoculate onto fresh Vero E6 monolayers. Overlay with Avicel-containing medium and incubate for 48-72 hours. Fix cells with formaldehyde and stain with crystal violet. Count plaques to calculate viral titer (PFU/mL) [5].
- Alternative Methods: TCIDâ‚…â‚€ assay or qRT-PCR for viral RNA quantification.
- Data Analysis: Calculate percentage viral titer reduction relative to untreated infected controls. Determine IC₅₀ values using non-linear regression analysis of dose-response data. A successful candidate, such as a PPNA targeting the 5'UTR, can demonstrate over 95% reduction in viral titer with an IC₅₀ of 0.8 μM [5].
Protocol 4: Non-Covalent Complexation of PNA with CPPs for Nuclear Delivery
This protocol describes the formation and optimization of non-covalent complexes between PNA and cell-penetrating peptides (CPPs) for efficient intracellular and nuclear delivery, comparing the efficacy of different CPPs like TP10, Tat, and TD2.2 [7].
Research Reagent Solutions
- PNA with NLS: PNA sequence (e.g., targeting ACLY or PCSK9) incorporating a C-terminal nuclear localization signal (NLS: -Pro-Lys-Lys-Lys-Arg-Lys-Val-amide) [7].
- CPP Solutions: TP10, Tat, and TD2.2 CPPs, each containing an NLS fragment. Synthesize via Fmoc-SPPS and purify by HPLC [7].
- Complexation Buffer: Serum-free cell culture medium or isotonic buffer.
Step-by-Step Procedure
- Complex Formation: Prepare separate solutions of PNA-NLS and each CPP in complexation buffer. Mix the PNA and CPP solutions at various molar ratios (e.g., 1:1, 1:5, 1:10 PNA:CPP) to optimize delivery efficiency. Vortex gently and incubate at room temperature for 15-30 minutes to allow complex formation [7].
- Cellular Uptake:
- Culture appropriate target cells (e.g., hepatocytes for PCSK9 targeting) in multi-well plates.
- Wash cells with PBS and replace medium with serum-free complexation buffer.
- Apply the PNA-CPP complexes to cells and incubate for 2-4 hours at 37°C.
- For fluorescently labeled PNA (Fluo-PNA), monitor uptake directly via flow cytometry or confocal microscopy [7].
- Efficiency Evaluation: Compare the delivery efficiency of different CPPs (TP10, Tat, TD2.2) by measuring cellular fluorescence or by evaluating the biological effect (e.g., downregulation of ACLY or PCSK9 mRNA/protein levels using qRT-PCR or Western blot, respectively). Studies indicate TP10 is often more effective than TD2.2 and Tat [7].
- Viability Assessment: Perform an MTT assay post-treatment to ensure the PNA-CPP complexes do not significantly impact cell viability at the working concentrations [7].
The Scientist's Toolkit: Key Reagents and Materials
Table 5: Essential Research Reagent Solutions for PNA Conjugate Work
| Category / Item | Critical Function | Application Notes |
|---|---|---|
| Fmoc-PNA Monomers | Core PNA backbone construction | Bhoc-protected nucleobases standard; specialty monomers (cpG, G+) available [3] |
| Functionalized Resins | Solid-phase synthesis support | Rink Amide, XAL-PEG-PS for C-terminal amide; TGA resin for peptide conjugation [7] [3] |
| Coupling Reagents | Activate monomers for amide bond formation | HATU/HOBt or PyAOP for manual/automated synthesis; DIC/DMAP for first residue [5] [3] |
| Cell-Penetrating Peptides | Enhance cellular delivery of PNA cargo | TP10, Tat, PolyArg, Transportan; efficacy varies (TP10 often superior) [26] [7] |
| Specialty Bases | Backbone & nucleobase modifications | Cyclopentane (cp) monomers, N7-methylguanine (G+) enhance brightness/affinity [3] |
| Fluorescent Tags | Tracking, detection, and diagnostics | FITC, NHS-fluorescein for labeling; BisQ as surrogate base in FIT-PNA sensors [25] [3] |
| Delivery Nanocarriers | Advanced in vivo delivery systems | PA3.2-HumAfFt, liposomes, polymer nanoparticles [25] |
Optimizing PNA Conjugates: Strategies for Synthesis, Purity, and Bioavailability
The synthesis of peptide-peptide nucleic acid (PNA) conjugates represents a cornerstone of modern antisense therapeutic development. These conjugates harness the high binding affinity and enzymatic stability of PNAs with the cellular delivery capabilities of peptide carriers. The linkage chemistry between these two modules is not merely a connective fixture but a critical determinant of the conjugate's solubility, stability, and ultimate biological efficacy. Within the context of automated platforms for peptide-PNA conjugate synthesis, the selection of an efficient, bio-orthogonal conjugation chemistry is paramount to leveraging the full potential of rapid, high-throughput production technologies. This Application Note details the implementation and advantages of Cysteine-2-Cyanoisonicotinamide (Cys-CINA) click chemistry alongside other prominent click-based linkages, providing researchers with validated protocols for their integration into automated synthesis workflows.
Click Chemistry Landscape for PNA Conjugation
The demand for efficient, selective, and biocompatible conjugation strategies has made click chemistry an indispensable tool. The table below compares three key click chemistries relevant for peptide-PNA conjugate synthesis.
Table 1: Comparison of Click Chemistries for Peptide-PNA Conjugation
| Conjugation Type | Reaction Partners | Key Advantages | Limitations | Typical Reaction Conditions |
|---|---|---|---|---|
| Cys-CINA [29] | N-terminal Cysteine & CINA | High selectivity; cost-effective; improved aqueous solubility; simple one-step reaction | Requires N-terminal cysteine | Aqueous/organic solvent, pH 7-8, room temperature, minutes to hours |
| Cys-CBT [30] [31] [29] | 1,2-Aminothiol & CBT | Extremely fast kinetics (~120x faster than CuAAC); bio-orthogonal; no metal catalyst; high efficiency | CBT derivatives can be expensive and hydrophobic, potentially compromising conjugate solubility | Physiological conditions (pH 7.4), room temperature |
| Copper-Catalyzed Azide-Alkyne (CuAAC) [32] | Azide & Alkyne | High specificity; well-established; reliable with diverse substrates | Requires cytotoxic copper catalyst; requires specialized ligands to prevent biomolecule degradation | Copper sulfate, sodium ascorbate, tris-triazolylamine ligand, room temperature |
Cys-CINA Conjugation: Protocol and Application
The Cys-CINA reaction is a bio-orthogonal ligation between an N-terminal cysteine residue and the nitrile group of a 2-cyanoisonicotinamide (CINA) derivative, resulting in the formation of a stable thiazole ring [29]. This linkage is less hydrophobic than the analogous CBT product, leading to improved aqueous solubility of the final conjugate—a critical property for biological activity [29].
Detailed Experimental Protocol
Objective: To conjugate a CINA-functionalized peptide to a PNA sequence bearing an N-terminal cysteine.
Table 2: Research Reagent Solutions for Cys-CINA Conjugation
| Item | Function/Description | Example & Specification |
|---|---|---|
| CINA Reagent | Electrophilic coupling partner | e.g., 2-Cyanoisonicotinic acid, >95% purity |
| N-terminal Cys-PNA | Nucleophilic coupling partner | PNA synthesized with N-terminal Cys residue |
| Coupling Reagents | Activates CINA carboxylic acid | HCTU, HATU, or DIC (3-4 equivalents) |
| Base | Facilitates activation and reaction | DIPEA or NMM (6-10 equivalents) |
| Solvent | Reaction medium | Anhydrous DMF, NMP, or aqueous buffer (pH ~7.5) |
| Purification System | Isolate final conjugate | RP-HPLC system with C18 column |
Procedure:
- Synthesis of CINA-functionalized Peptide: Synthesize the cell-penetrating peptide (CPP) or antimicrobial peptide on solid support using standard Fmoc-SPPS. Incorporate a CINA moiety at the C-terminus by coupling 2-cyanoisonicotinic acid (4 equivalents) using standard activation with HCTU (3.9 equiv) and DIPEA (6 equiv) in DMF for 30-60 minutes [29].
- Synthesis of N-terminal Cys-PNA: Synthesize the antisense PNA sequence on Rink amide resin. Following the final Fmoc deprotection, couple an 8-amino-3,6-dioxaoctanoic acid (PEG) spacer, followed by an Fmoc-Cys(Trt)-OH. Perform a final Fmoc deprotection to reveal the N-terminal cysteine [29].
- Cleavage and Purification: Cleave the CINA-peptide and Cys-PNA from their respective resins using standard cleavage cocktails (e.g., TFA:Water:TIPS, 95:2.5:2.5). Precipitate the crude products in cold diethyl ether, purify via reversed-phase HPLC, and confirm identity by LC-MS.
- Conjugation Reaction:
- Dissolve the purified CINA-peptide (1.0 equivalent) and Cys-PNA (1.2 equivalents) in a suitable solvent (e.g., 50 mM phosphate buffer, pH 7.5, or a mixture of water and acetonitrile).
- Adjust the pH to 7.5-8.0 if necessary.
- Allow the reaction to proceed at room temperature with gentle agitation. Monitor reaction completion by analytical LC-MS (typically 1-4 hours).
- Purification of Final Conjugate: Purify the crude peptide-PNA conjugate using semi-preparative reversed-phase HPLC. Lyophilize the pure fractions to obtain the final conjugate as a solid for downstream biological evaluation.
Application in Antimicrobial Conjugate Development
This Cys-CINA protocol has been successfully applied to create antimicrobial conjugates. For instance, a PNA targeting the essential acpP gene was conjugated to the membrane-active peptide polymyxin B. The resulting conjugate exhibited enhanced antibacterial activity against multidrug-resistant Acinetobacter baumannii compared to the PNA alone, validating the Cys-CINA linkage as an effective strategy for developing novel antisense antimicrobials [29].
Diagram 1: Cys-CINA conjugation workflow for generating peptide-PNA conjugates.
Alternative Click Conjugation Strategies
The CBT-Cys Click Reaction
The condensation between a 1,2-aminothiol (e.g., from an N-terminal cysteine) and 2-cyanobenzothiazole (CBT) is a biocompatible reaction inspired by firefly luciferin biosynthesis [30] [31]. This reaction proceeds efficiently at physiological pH without metal catalysts.
Key Protocol Considerations:
- Reaction Setup: The CBT derivative (e.g., functionalized with a biotin or fluorescent dye) is incubated with a peptide or protein containing an N-terminal cysteine in phosphate-buffered saline (PBS) at pH 7.4 [30] [31].
- Kinetics: The reaction is notably fast, with a second-order rate constant of approximately 9.1 Mâ»Â¹sâ»Â¹, facilitating rapid conjugation [31].
- Applications: Beyond simple labeling, the CBT-Cys reaction enables the controllable self-assembly of nanoparticles for imaging and drug delivery, leveraging the formation of CBT-dimers that self-assemble under specific cellular conditions [31].
Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC)
The CuAAC reaction between an azide and a terminal alkyne to form a 1,2,3-triazole linker remains a powerful and widely used method, particularly for templated ligations [32].
Key Protocol Considerations:
- Template-Directed Ligation: For PNA-DNA conjugation, a DNA template can be used to co-localize a 5'-azide-modified DNA with a 3'-alkyne-modified PNA. This strategy enables highly efficient and sequence-specific ligation with single-nucleotide discrimination capability [32].
- Critical Optimization: The copper(I) catalyst, often generated in situ from copper(II) sulfate and sodium ascorbate, can cause significant degradation of nucleic acids. The addition of a tris-triazolylamine ligand (THPTA) is essential to suppress this degradation and achieve near-quantitative yields of the conjugation product [32].
- Standard Reaction Conditions: A typical 50 µL reaction mixture contains 50 nM azide-modified DNA, equimolar alkyne-modified PNA, 100 mM NaCl, 10 mM potassium phosphate buffer, 1 mM CuSO₄, 1 mM THPTA, and 5 mM sodium ascorbate. The reaction proceeds to completion within 1 hour at room temperature [32].
Diagram 2: Two primary click chemistry strategies for bioconjugation, highlighting key reaction requirements.
The integration of advanced click chemistries like Cys-CINA, CBT-Cys, and CuAAC into automated synthesis platforms provides a powerful foundation for the rapid generation of peptide-PNA conjugate libraries. The choice of chemistry involves a strategic balance between factors such as solubility, kinetics, catalyst requirements, and final application. The Cys-CINA reaction, with its optimal balance of selectivity, cost, and favorable physicochemical properties for bioactive conjugates, represents a particularly valuable tool for researchers developing next-generation antisense therapeutics, especially in the antimicrobial domain.
Within the development of an automated platform for peptide-peptide nucleic acid (PNA) conjugate synthesis, optimizing the physicochemical properties of PNA oligomers is a fundamental research challenge. PNAs, with their neutral pseudopeptide backbone, often suffer from limited aqueous solubility and a tendency to aggregate, which can complicate synthesis, purification, and handling [33]. Furthermore, while their inherent affinity for complementary DNA and RNA is high, there is significant scope to enhance binding strength and sequence specificity for demanding applications such as double-stranded DNA invasion [34]. This Application Note details proven chemical strategies for modifying the PNA backbone and sequence to simultaneously improve aqueous solubility and target affinity. The protocols herein are designed to be compatible with automated solid-phase synthesis platforms, enabling the robust production of high-performance PNA conjugates for therapeutic and diagnostic development.
Strategic Approaches to Modification
The optimization of PNA properties can be achieved through two primary, complementary approaches: terminal sequence modifications and backbone alterations. Table 1 summarizes the key modification strategies, their primary benefits, and their implementation.
Table 1: Strategic Modifications for Enhancing PNA Solubility and Affinity
| Modification Strategy | Key Benefit(s) | Typical Implementation | Compatibility with Automation |
|---|---|---|---|
| Terminal Lysine Residues | Improves aqueous solubility via introduction of a positive charge [33] [35]. | Addition of 1-2 lysines at C-terminus. | High; uses standard Fmoc-chemistry. |
| O Linkers (AEEA) | Enhances solubility; acts as a spacer for conjugates [35]. | Incorporation of 1-2 linkers at sequence ends. | High; available as standard building blocks. |
| γ-Backbone Modification | Pre-organizes backbone, significantly improving binding affinity and cellular uptake [34] [33]. | Substitution of standard monomer with γ-modified monomers (e.g., miniPEG, guanidine). | Moderate; requires specialized monomers. |
| Cationic Peptide Conjugation | Dramatically improves cellular delivery and can enhance solubility [36] [34]. | Conjugation of oligolysine or arginine-rich peptides post-synthesis or during synthesis. | High for co-synthesis; standard for click chemistry. |
The following diagram illustrates the logical decision-making process for selecting and combining these modification strategies to achieve desired PNA properties.
Detailed Modification Protocols
Protocol 1: Incorporation of Solubility-Enhancing Termini
This protocol describes the co-synthesis of terminal lysine residues and O-linkers during automated solid-phase PNA synthesis to significantly improve aqueous solubility [33] [35].
- Objective: To increase the aqueous solubility of a PNA oligomer by introducing charged and hydrophilic groups at its C-terminus.
- Principle: The addition of positively charged lysine residues or hydrophilic O-linkers (8-amino-3,6-dioxaoctanoic acid, AEEA) disrupts PNA self-aggregation and improves solvation in aqueous buffers [35].
- Materials:
- Workflow:
- Key Notes:
- This modification is particularly critical for purine-rich sequences, which have a higher tendency to aggregate [33].
- The use of low-loading Rink Amide Resin is recommended to minimize intermolecular aggregation during synthesis [33].
- The C-terminal -CONH2 from the Rink Amide Resin also contributes to solubility.
Protocol 2: Cys-CINA Conjugation for Peptide-PNA Conjugate Synthesis
This protocol outlines the use of Cysteine-2-Cyanoisonicotinamide (Cys-CINA) click chemistry for linking PNAs to cell-penetrating peptides (CPPs), a method that improves conjugation efficiency and conjugate solubility compared to other strategies [36].
- Objective: To create a stable, soluble conjugate between a PNA strand and a delivery peptide (e.g., a CPP or antimicrobial peptide like polymyxin).
- Principle: The reaction between a PNA with an N-terminal cysteine and a peptide functionalized with 2-cyanoisonicotinamide (CINA) forms a stable thiazole ring linkage. This method is highly selective, proceeds in high yield, and results in a less hydrophobic conjugate compared to similar methods like Cys-CBT [36].
- Materials:
- PNA with N-terminal cysteine (Cys-PNA), synthesized per standard protocols.
- Peptide modified with 2-cyanoisonicotinamide (CINA) group.
- Ammonium acetate buffer (0.1 M, pH 4.5) or DMF/water mixture.
- LC-MS and RP-HPLC systems for analysis and purification.
- Workflow:
- Key Notes:
The Scientist's Toolkit: Essential Research Reagents
The successful implementation of the above protocols relies on a set of key reagents and instruments. Table 2 catalogs these essential components.
Table 2: Key Reagents and Instruments for PNA Modification and Synthesis
| Item | Function / Purpose | Implementation Example / Note |
|---|---|---|
| Fmoc-PNA(Bhoc)-OH Monomers | Building blocks for standard solid-phase PNA synthesis [36] [33]. | Used with HCTU/DIPEA activation in NMP [36]. |
| Fmoc-Lys(Boc)-OH | Introduces a primary amine for solubility or conjugation. Added at C-terminus to improve solubility [33] [35]. | |
| Fmoc-8-amino-3,6-dioxaoctanoic acid (O-Linker) | Polyethylene glycol-based spacer that enhances solubility and provides a spacer between PNA and labels/peptides [36] [35]. | |
| γ-Modified PNA Monomers | Pre-organizes PNA backbone into a right-handed helix, dramatically improving binding affinity to DNA/RNA [34] [33]. | γ-(S)-miniPEG PNA is a prominent example that also improves solubility. |
| CINA Reagent (2-Cyanoisonicotinamide) | Enables efficient, hydrophilic conjugation of peptides to PNAs via click chemistry with an N-terminal cysteine [36]. | Superior to cyanobenzothiazole (CBT) in cost and hydrophobicity. |
| Rink Amide Resin | Solid support for PNA synthesis, providing a C-terminal amide upon cleavage [36] [5]. | Low-loading resin (e.g., 0.18-0.5 mmol/g) is preferred to reduce aggregation [5] [33]. |
| Automated Synthesizer | Enables rapid, reproducible synthesis of PNA and peptide-PNA conjugates. | Platforms like the MultiPep 2 (for parallel synthesis) [37] or custom flow synthesizers [5] are used. |
Concluding Remarks
The strategic modification of PNA through terminal functionalization and backbone engineering is essential for transforming these high-affinity oligonucleotide mimics into practical tools for research and therapy. The protocols detailed in this application note—incorporating lysine residues, O-linkers, and employing the Cys-CINA conjugation chemistry—provide a robust and actionable framework for significantly enhancing the aqueous solubility and target affinity of PNA oligomers. When integrated into an automated synthesis platform, these strategies enable the high-throughput production of well-characterized, high-performance PNA conjugates, accelerating their path toward diagnostic and therapeutic application.
On-Resin Cyclization and Late-Stage Functionalization for Enhanced Stability
Application Notes
The development of automated, high-purity synthesis platforms represents a transformative advancement in the production of cyclic peptide-peptide nucleic acid (PNA) conjugates. These platforms address critical limitations in traditional synthesis methods, enabling robust and scalable access to complex architectures with enhanced stability and binding properties. The integration of on-resin head-to-tail cyclization with fully automated fast-flow chemistry has successfully accelerated production by an order of magnitude while achieving remarkable crude purity levels of up to 95% [4] [27]. This technological progress is pivotal for drug discovery and nanotechnology applications, where the remarkable nuclease resistance and high binding affinity of cyclic PNAs can be fully exploited [4] [1].
The broader context of this research aligns with the pressing need for synthetic methodologies that bridge the gap between conventional oligonucleotide and peptide synthesis. Peptide Nucleic Acids (PNAs) are synthetic DNA/RNA mimics where the sugar-phosphate backbone is replaced by repeating N-2-aminoethyl glycine units, conferring superior binding affinity, specificity, and enzymatic resistance compared to natural oligonucleotides [1]. However, their adoption has been hampered by inefficient synthesis strategies and inherent challenges in cellular delivery [5] [25]. The automated platform for peptide-PNA conjugate synthesis directly addresses these limitations, facilitating the systematic investigation of structure-function relationships and accelerating the development of PNA-based therapeutics [5].
Quantitative Performance Data
The performance of the automated synthesis platform is demonstrated through the construction and screening of diverse PNA/PPNA libraries. The following table summarizes key quantitative data from these studies.
Table 1: Performance Metrics of Automated Cyclic PNA/PPNA Synthesis and Screening
| Parameter | Performance Metric | Experimental Context |
|---|---|---|
| Crude Purity | Up to 95% [4] [27] [6] | 2- to 12-residue cyclic PNA/PPNA sequences synthesized using the iChemAFS platform with DAN linker. |
| Synthesis Time Reduction | Order of magnitude (10x) faster [4] [27] | Comparison of automated fast-flow versus manual synthesis approaches. |
| Amide Bond Formation Rate | 10 seconds per bond [5] | Achieved using high-temperature (70°C) flow chemistry on the automated microscale flow synthesizer. |
| Library Size | 22 structures [4] [27] | Library of PNA/PPNA constructs built and screened for membrane permeability. |
| Binding Affinity Improvement | 30-fold to 280-fold [38] | Improvement for PNA-linker probes compared to non-functional PEG linkers in RNA binding assays. |
| Antiviral Efficacy | >95% viral titer reduction (IC50 = 0.8 μM) [5] | PPNA targeting SARS-CoV-2 5'UTR in a live virus infection assay. |
The data underscore the efficiency and effectiveness of the automated platform. The combination of high crude purity and dramatically reduced synthesis times enables the rapid generation of compound libraries for biological evaluation. Furthermore, the functional outcomes, such as significant enhancements in binding affinity and potent antiviral activity, validate the platform's utility for developing bioactive molecules [38] [5].
Biological Efficacy and Functional Enhancement
The application of this synthetic methodology has yielded conjugates with significant biological efficacy. In live virus infection assays, a PPNA designed to target the 5' untranslated region (5'UTR) of SARS-CoV-2 genomic RNA achieved a reduction in viral titer by over 95%, demonstrating the potential of these conjugates as antiviral agents [5]. Beyond antivirals, the technology facilitates the synthesis of complex architectures such as cyclic PPNA nanotubes with high aspect ratios, characterized by TEM and AFM, highlighting applications in nanotechnology and material science [4] [27].
A key advantage of cyclic PNA/PPNAs is their enhanced stability and cellular permeability. Screening of the synthesized library using parallel artificial membrane permeability assays (PAMPA) confirmed promising membrane permeability profiles [4]. This is crucial for therapeutic applications, as the neutral polyamide backbone of PNAs, while conferring nuclease resistance, often results in poor cellular uptake [1] [25]. The automated platform allows for the straightforward incorporation of cell-penetrating peptides (CPPs) to further ameliorate this challenge, improving solubility and promoting cellular internalization [5].
Experimental Protocols
Automated On-Resin Head-to-Tail Cyclization of PNA/PPNAs
This protocol describes the automated synthesis and cyclization of peptide-PNA conjugates using a diaminonicotinic acid (DAN) linker on the iChemAFS platform, enabling the production of cyclic structures with up to 95% crude purity [4] [27] [6].
Materials and Reagents
- Solid Support: Rink Amide resin (0.5 mmol/g loading).
- PNA Monomers: Fmoc-protected PNA monomers with standard nucleobases (A, C, G, T).
- Linker: Fmoc-diaminonicotinic acid (DAN) linker.
- Activation Reagents: PyAOP ((7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate).
- Base: N,N-Diisopropylethylamine (DIEA).
- Solvent: Anhydrous N,N-Dimethylformamide (DMF).
- Deprotection Reagent: 20% (v/v) Piperidine in DMF.
- Cleavage Reagent: Trifluoroacetic Acid (TFA)-based cocktail.
Equipment
- iChemAFS or equivalent automated fast-flow synthesizer.
- HPLC system with reverse-phase C-18 column for analysis and purification.
- High-resolution mass spectrometer (HRMS with MALDI or TOF) for characterization.
Step-by-Step Procedure
Resin Loading and Initial Setup:
- Load approximately 15 mg of Rink Amide resin (0.5 mmol/g) into the reusable reactor body of the flow synthesizer. The system is designed for a 7.5 μmol scale synthesis [5].
- Swell the resin in DMF for a minimum of 30 minutes prior to commencing synthesis.
Chain Elongation (Performed Automatically by the iChemAFS Platform):
- Deprotection: Flush the resin-bound growing chain with a solution of 20% piperidine in DMF at a flow rate of 2.5 mL/min. The system merges the reagent with a pre-heated zone to achieve an efficient deprotection temperature of ~40°C, removing the Fmoc group. This step is monitored in-line by UV-vis to track Fmoc-removal [5].
- Coupling: Simultaneously draw solutions of the incoming Fmoc-PNA monomer (10 equiv), activator PyAOP (9.6 equiv), and base DIEA (30 equiv) in DMF from the storage system.
- Merge the streams and pass the mixture through a heating element set to 70°C to form the activated ester.
- Direct the activated solution through the resin bed, also maintained at 70°C, for 10 seconds to complete the amide bond formation [5].
- Wash the resin with DMF to remove excess reagents.
- Repeat the deprotection and coupling cycles until the full linear sequence is assembled.
On-Resin Cyclization using the DAN Linker:
- After assembly of the linear sequence, incorporate the DAN linker at the N-terminus following the standard coupling procedure.
- Following Fmoc deprotection of the DAN linker, the free amine is ready for cyclization.
- The cyclization is initiated by the automated system using the same coupling chemistry (PyAOP/DIEA, 70°C) to form an amide bond between the N-terminal amine of the DAN linker and the C-terminal carboxylic acid of the resin-bound conjugate, resulting in head-to-tail cyclization [4] [27].
Cleavage and Deprotection:
- Once cyclization is complete, cleave the cyclic conjugate from the resin and remove any remaining side-chain protecting groups using a TFA-based cleavage cocktail.
- Precipitate the crude product in cold diethyl ether.
- Centrifuge and isolate the pellet.
Purification and Analysis:
- Purify the crude cyclic PNA/PPNA using reverse-phase HPLC.
- Analyze the final product and confirm its identity by HRMS [1].
Diagram: Automated Fast-Flow Synthesis and Cyclization Workflow
Late-Stage Serine Modification via Deoxygenative Functionalization
This protocol enables the site-selective transformation of serine residues within complex peptides into various noncanonical amino acids, such as homoglutamine and phosphonates, via carbon-carbon bond formation. It is compatible with peptides on solid support or in solution, making it ideal for diversifying cyclic PNA-peptide conjugates after their primary structure is established [39].
Materials and Reagents
- Phosphoramidite Reagent: Phosphoramidite 1 (bearing a tethered aryl iodide).
- Activator: 5-Methyl-tetrazole.
- Photocatalyst: Photocatalyst 6 (e.g., Ir-based) or 10-Phenylphenothiazine (PTH).
- Hydrogen Atom Donor: IDM-BH3 (IDM = dimethyl imidazolylidene).
- Additive: Potassium Formate (HCO2K).
- Radical Acceptors: e.g., Vinyl nitrile, vinyl phosphate, acrylamides, acryl esters.
- Solvents: Anhydrous acetonitrile (MeCN), DMF.
Equipment
- Schlenk flasks or glass vials for reactions under inert atmosphere.
- Photoreactor or blue LED light source (e.g., 34 W Kessil lamp).
- Rotary evaporator.
- HPLC system for purification.
Step-by-Step Procedure
Phosphitylation of Serine Residue:
- Dissolve the peptide containing a free serine side chain (on-resin or in solution, 1.0 equiv) in anhydrous MeCN in a reaction vessel suitable for irradiation.
- Add phosphoramidite 1 (2.0 equiv) and 5-methyl-tetrazole (4.0 equiv) under an inert nitrogen atmosphere.
- Stir the reaction mixture at room temperature for 1-2 hours to form the phosphite intermediate 3, which can be isolated in up to 92% yield [39].
Photocatalytic Radical Generation and Giese Addition:
- To the same reaction vessel containing the phosphite intermediate 3, add the chosen radical acceptor (e.g., vinyl nitrile, 5.0 equiv), photocatalyst 6 (2 mol%), IDM-BH3 (2.0 equiv), and HCO2K (2.0 equiv).
- Seal the vessel and degas the reaction mixture by purging with nitrogen for 10-15 minutes to remove oxygen, which quenches radical intermediates.
- Irradiate the reaction mixture with blue LED light while stirring vigorously for 6-16 hours at room temperature.
Reaction Work-up:
- After completion (monitored by LC-MS), concentrate the reaction mixture under reduced pressure.
- If the reaction was performed on-resin, wash the resin thoroughly with DMF and MeCN before proceeding with standard cleavage.
- If performed in solution, purify the crude product directly by reverse-phase HPLC to isolate the modified peptide.
Product Analysis:
- Characterize the final product by HRMS to confirm identity and chiral HPLC to verify the absence of epimerization at the α-carbon [39].
Diagram: Mechanism of Late-Stage Serine Modification
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents and materials essential for implementing the described protocols for on-resin cyclization and late-stage functionalization.
Table 2: Essential Research Reagents for PNA Conjugate Synthesis and Modification
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Diaminonicotinic Acid (DAN) Linker | Facilitates on-resin head-to-tail cyclization by providing a structural scaffold for ring closure. | Enables precise control over cyclization, achieving high crude purity (up to 95%) for 2-12 residue sequences [4] [27]. |
| PyAOP Activator | Coupling reagent for efficient amide bond formation between PNA monomers. | Used in the automated flow synthesizer at 70°C to achieve ultra-fast coupling (10 seconds/bond) without need for capping [5]. |
| Phosphoramidite Reagent 1 | Activates serine side-chain hydroxyl groups for deoxygenative radical generation. | Key to late-stage functionalization. Contains a tethered aryl iodide for radical cyclization and β-scission [39]. |
| IDM-BH3 (IDM-Borane) | Serves as a critical hydrogen atom donor in the photocatalytic cycle. | Terminates the radical cascade, directly incorporating a hydrogen (or deuterium from IDM-BD3) into the final product [39]. |
| Rink Amide Resin | Solid support for synthesis. Provides a amide handle upon cleavage for C-terminal amide conjugates. | Standard loading of 0.5 mmol/g is used in the automated flow reactor (15 mg for 7.5 μmol scale) [5] [25]. |
| Cell-Penetrating Peptides (CPPs) | Conjugated to PNAs to improve cellular uptake and solubility. | Covalent attachment creates PPNAs, crucial for biological activity in live cells (e.g., antisense applications) [5]. |
Within the development of an automated platform for peptide-PNA conjugate (PPNA) research, achieving high crude purity is a fundamental challenge. Synthesis pitfalls, particularly the formation of deletion sequences and nucleobase adducts, significantly compromise product yield and efficacy, hindering rapid progression in therapeutic and diagnostic applications [5]. These by-products arise from inefficient coupling and depurination side reactions during solid-phase synthesis. This Application Note details optimized protocols and quantitative data demonstrating how automated fast-flow synthesis mitigates these specific issues, enabling the production of long PNA conjugates with superior crude purity for drug discovery and development.
Quantitative Analysis of Synthesis Pitfalls and Optimization
In-line UV-vis monitoring combined with LC-MS and HPLC characterization enables rapid identification and quantification of major synthesis impurities. The following table summarizes the performance of different synthesis conditions for a model 4-mer PNA sequence (ACTG-Gly-CONHâ‚‚), clearly illustrating the impact of method and temperature on purity [5].
Table 1: Evaluation of Reaction Conditions for Automated Flow PNA Synthesis and Its Impact on Purity [5]
| Entry | Synthesis Method | Reaction Temperature | Crude Purity (%) | Total Deletions (%) | Nucleobase Adducts (%) | Key Condition Modifications |
|---|---|---|---|---|---|---|
| 1 | Manual Batch | Not specified | 57 | ~4 | ~2 | Standard protocols with 6 equiv monomers [5] |
| 2 | Automated Flow | 70°C | 70 | 10 | 7 | Initial high-temperature acceleration |
| 3 | Automated Flow | 70°C | 68 | 9 | 8 | Base screening: Piperazine |
| 4 | Automated Flow | 70°C | 75 | 5 | 3 | Base screening: Morpholine |
| 5 (Optimal) | Automated Flow | 70°C (Coupling) / ~40°C (Deprotection) | >90 | <1 | <1 | Optimal: Piperidine, 10 equiv monomers, 9.6 equiv PyAOP, 30 equiv DIEA |
The data demonstrates that the optimized automated flow protocol (Entry 5) reduces deletion sequences to below 1% and minimizes nucleobase adduct formation, achieving a crude purity exceeding 90%. This is a marked improvement over both standard manual synthesis and non-optimized flow conditions.
Automated Fast-Flow Synthesis Protocol
This protocol is designed for a fully automated flow instrument (e.g., iChemAFS platform) and is scalable for 7.5 μmol synthesis using 15 mg of Rink Amide resin (0.5 mmol/g) [5] [27].
Key Reagents and Materials
Table 2: Research Reagent Solutions for Automated Flow PNA Synthesis
| Reagent / Material | Function / Role | Specification / Comment |
|---|---|---|
| Rink Amide Resin | Solid support for synthesis | Loading: 0.5 mmol/g [5] |
| Fmoc-PNA Monomers | Building blocks for chain elongation | 10 equivalents per coupling cycle [5] |
| PyAOP Activator | Activates PNA monomers for coupling | 9.6 equivalents per cycle [5] |
| DIEA (Base) | Base for coupling reaction | 30 equivalents in DMF [5] |
| Piperidine Solution | Removal of Fmoc protecting group | Optimal base for high-temperature deprotection [5] |
| DMF Solvent | Primary reaction solvent | Stored under nitrogen atmosphere [5] |
Step-by-Step Workflow
The automated synthesis cycle, managed by a modular script in the Mechwolf programming environment, is completed within 3 minutes per monomer addition [5].
Figure 1: Workflow of an automated fast-flow PNA synthesis cycle. The process alternates between high-temperature coupling and controlled-temperature deprotection, with in-line monitoring [5].
Coupling Step
- Reagent Merging: Three HPLC pumps simultaneously draw solutions of the PNA monomer (10 equiv), activator PyAOP (9.6 equiv), and base DIEA (30 equiv) from a nitrogen-atmosphere storage system [5].
- Activation and Reaction: The solutions are merged via a valve and passed through a module heated to 70°C, forming the activated ester. This mixture immediately flows through the packed bed of resin, also maintained at 70°C [5].
- Bond Formation: The amide bond formation is completed within 10 seconds at this elevated temperature. No capping step is required due to the high coupling efficiency [5].
Deprotection Step
- Controlled Environment: A piperidine solution is pumped through a room-temperature loop, which then meets the 70°C reactor. This creates a controlled deprotection environment of approximately 40°C [5].
- Efficiency and Safety: This temperature is optimal for rapid and efficient removal of the Fmoc protecting group while minimizing the formation of nucleobase adducts, particularly aspartimides [5].
- Process Monitoring: An in-line UV-vis detector monitors the composition of the spent reagent solution, specifically tracking the Fmoc-removal absorbance chromatogram to infer deprotection efficiency and mass transfer rates [5].
Discussion
Mechanistic Insights into Pitfall Prevention
The automated fast-flow protocol directly addresses the root causes of common synthesis pitfalls:
- Preventing Deletion Sequences: The combination of a high temperature (70°C) and a large excess of reagents (monomer, activator, base) dramatically accelerates coupling kinetics and ensures reaction completion within seconds. This eliminates the need for double-couplings and minimizes truncated sequences resulting from inefficient reactions [5].
- Minimizing Nucleobase Adducts: The strategic use of a lower deprotection temperature (~40°C) is critical. While high temperature accelerates coupling, it can promote side reactions during the base-sensitive deprotection step. The segmented temperature control protects nucleobases from degradation and adduct formation, such as piperidine adducts, while maintaining high efficiency [5]. The choice of piperidine as the deprotection base was also identified as optimal through systematic screening [5].
Application in Conjugate Synthesis
The reliability of this optimized method enables the direct, "single-shot" synthesis of long PNA sequences conjugated to cell-penetrating peptides (CPPs), which are crucial for therapeutic delivery [5]. This technology has been successfully used to synthesize an 18-mer PPNA for β-thalassemia splice-correction and eight anti-SARS-CoV-2 PPNAs in a single day, demonstrating its utility in rapid drug candidate screening [5]. The high crude purity (>90%) simplifies downstream purification and is essential for obtaining accurate biological readouts in structure-activity relationship studies.
This Application Note provides a validated, detailed protocol for an automated fast-flow synthesizer that effectively mitigates the critical synthesis pitfalls of deletion sequences and nucleobase adducts in PNA synthesis. By implementing high-temperature coupling with segmented temperature control for deprotection, researchers can achieve high crude purity and significantly accelerate the production of peptide-PNA conjugates for drug discovery and development. This robust approach is a cornerstone for a reliable automated platform for PPNA research.
Proof of Concept: Validating PPNA Efficacy in Biological Systems
The urgent need for innovative antiviral agents has been underscored by the continuous emergence of SARS-CoV-2 variants and the limitations of current therapeutic options. Peptide Nucleic Acids (PNAs) represent a promising class of antisense oligonucleotides that combine strong RNA binding affinity with superior stability, yet their clinical translation has been hampered by poor cellular uptake [5]. The conjugation of PNAs to cell-penetrating peptides (CPPs) creates PPNAs, which address this critical delivery challenge [5] [40].
This case study details the application of a fully automated, high-throughput synthesis platform to produce anti-SARS-CoV-2 PPNAs. We present a comprehensive protocol, from automated conjugate synthesis to biological validation, which culminated in a candidate achieving over 95% reduction in viral titer in a live virus infection assay [5]. The methodology and data presented herein serve as a model for the rapid development of antisense therapeutics within the broader context of automated platform for peptide-PNA conjugate synthesis research.
Automated Flow Synthesis of PPNAs
Protocol: Fully Automated Single-Shot Synthesis
The synthesis of PPNA conjugates was achieved using a fully automated fast-flow instrument, which significantly accelerates production and improves crude purity compared to standard batch protocols [5].
- Instrument Setup: The automated flow PNA synthesizer comprises seven modules: a central control computer, a solution storage system, three HPLC pumps, three multiposition valves, heating elements, a reaction zone, and an in-line UV-vis detector. The entire process is controlled by a modular script in the Mechwolf programming environment [5].
- Reactor Specifications: A reusable reactor body is designed for a 7.5 μmol scale synthesis, typically loaded with 15 mg of Rink Amide resin (0.5 mmol/g). This microscale approach reduces expensive monomer consumption while delivering milligram quantities of product sufficient for biological characterization [5].
- Synthesis Cycle:
- Coupling: Three HPLC pumps deliver the PNA monomer, activator (PyAOP), and base (DIEA) solutions. These are merged and passed through a 70°C heating element to form an activated ester, which then flows through the resin-packed reactor at 70°C. Amide bond formation is completed within 10 seconds [5].
- Deprotection: A piperidine solution flows through the reactor to remove the Fmoc protecting group. The in-line UV-vis detector monitors the deprotection efficiency [5].
- Key Advantage: The entire solid-phase PPNA synthesis cycle is completed in approximately 3 minutes per monomer, an order of magnitude faster than commercial microwave peptide synthesizers [5]. This high-temperature flow synthesis eliminates the need for capping steps and prevents on-resin aggregation, enabling the direct synthesis of long (>15-mer) PNA sequences conjugated to peptides in a single, uninterrupted run [5].
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Essential Reagents and Materials for Automated PPNA Synthesis
| Reagent/Material | Function/Description |
|---|---|
| Rink Amide Resin | Solid support for synthesis (0.5 mmol/g loading) [5]. |
| Fmoc-PNA Monomers | Building blocks for PNA chain assembly (Bhoc-protected nucleobases) [5]. |
| PyAOP Activator | (7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate; activates monomer for coupling [5]. |
| DIEA Base | N,N-Diisopropylethylamine; used as a base in the coupling reaction [5]. |
| Piperidine Solution | Removes the Fmoc (Fluorenylmethyloxycarbonyl) protecting group after each coupling cycle [5]. |
| Cell-Penetrating Peptide (CPP) | Covalently attached to PNA to enhance cellular uptake and solubility (e.g., a 12-mer CPP was used in the featured study) [5]. |
Design and Antiviral Screening of Anti-SARS-CoV-2 PPNAs
Experimental Protocol: Design, Synthesis, and Screening
- PPNA Design: Eight PPNAs were designed to target different regions of the SARS-CoV-2 genomic RNA. The PNA sequence was an 18-mer, chosen for its potential for higher affinity and lower off-target effects, and was covalently linked to a 12-mer CPP [5].
- Synthesis Campaign: Utilizing the automated flow synthesizer, all eight target-specific PPNA candidates were manufactured within a single day, demonstrating the platform's capability for rapid library production [5].
- In Vitro Efficacy Screening:
- Cell-based Assay: The antiviral activity of the synthesized PPNAs was evaluated in a live virus infection assay using SARS-CoV-2.
- Outcome Measurement: Viral titer was measured post-treatment to quantify the antiviral effect. The most potent candidate, a PPNA targeting the 5' untranslated region (5'UTR) of the SARS-CoV-2 genomic RNA, reduced the viral titer by over 95% [5].
- Potency Quantification: The half-maximal inhibitory concentration (IC₅₀) for this leading PPNA candidate was determined to be 0.8 μM, indicating high potency [5].
Data Presentation: Antiviral Screening Results
Table 2: Key Experimental Data from the Anti-SARS-CoV-2 PPNA Study
| Parameter | Result | Context & Significance |
|---|---|---|
| Viral Titer Reduction | > 95% | Observed with the lead PPNA candidate in a live SARS-CoV-2 infection assay [5]. |
| IC₅₀ Value | 0.8 μM | The concentration needed to inhibit 50% of viral activity, demonstrating high potency [5]. |
| PNA Sequence Length | 18-mer | Longer sequences can offer higher affinity and specificity for the RNA target [5]. |
| CPP Length | 12-mer | The cell-penetrating peptide component crucial for cellular internalization [5]. |
| Synthesis Time for 8 PPNAs | 1 day | Highlights the exceptional throughput of the automated flow synthesis platform [5]. |
| Coupling Time per Monomer | 10 seconds | Drastic reduction compared to the 10+ minutes required by conventional synthesizers [5]. |
Workflow and Mechanism Analysis
Diagram: Automated PPNA Synthesis and Screening Workflow
The following diagram illustrates the integrated process from automated synthesis to antiviral validation, as detailed in the protocols.
Diagram 1: PPNA Development Workflow.
Diagram: Proposed Antiviral Mechanism of PPNAs
The antiviral activity of the lead PPNA is achieved through a steric blockade mechanism, as visualized below.
Diagram 2: PPNA Antiviral Mechanism.
This case study demonstrates a successful, integrated application of automated synthesis to a pressing drug discovery challenge. The ability to rapidly produce a library of PPNAs and identify a candidate with exceptional antiviral efficacy against SARS-CoV-2 validates the automated platform as a powerful tool for accelerating the development of oligonucleotide-based therapeutics. The lead PPNA candidate, characterized by its high affinity (18-mer PNA), efficient delivery (12-mer CPP), and potent antiviral effect (>95% viral titer reduction, IC₅₀ = 0.8 μM), offers a promising strategy for COVID-19 treatment and establishes a robust workflow for responding to future viral threats.
The escalating global antimicrobial resistance (AMR) crisis demands innovative therapeutic strategies that can overcome conventional antibiotic mechanisms. Pathogen genomics has revolutionized our understanding of bacterial resistance, providing deep insights into the mechanisms and dissemination of AMR by identifying essential genes critical for bacterial survival [41]. These essential genes represent promising targets for novel antimicrobial approaches [42]. Concurrently, Peptide Nucleic Acids (PNAs) have emerged as powerful antisense agents capable of modulating gene expression through sequence-specific binding to complementary DNA or RNA [1]. When conjugated to cell-penetrating peptides (CPPs) to form peptide-PNA conjugates (PPNAs), these molecules gain significantly improved cellular uptake and target specificity [43] [5]. This case study explores the integration of an automated synthesis platform for developing antimicrobial PPNAs targeting essential genes in resistant bacteria, framed within broader thesis research on automated PPNA synthesis platforms.
Background and Significance
The Antimicrobial Resistance Crisis
Antimicrobial resistance poses a critical global public health threat, with bacterial pathogens of primary concern. By 2050, AMR is estimated to cause approximately 10 million annual deaths with economic losses totaling $1.7 trillion based on disability-adjusted life years lost [41]. The Gram-negative pathogen Acinetobacter baumannii exemplifies this threat, categorized as an 'urgent threat' due to certain clinical strains having developed resistance to all known therapeutics [42]. The One Health framework—recognizing the interconnectedness of human, animal, and environmental health—is crucial for understanding AMR transmission and evolution across different sectors [41].
Essential Genes as Therapeutic Targets
Essential genes, which are vital for bacterial survival even under optimal growth conditions, represent promising targets for drug discovery [42]. In A. baumannii, essential genes have been cataloged using methods like transposon sequencing (Tn-seq), which identifies genes with low or nonexistent insertion frequencies as critical for survival [42]. However, traditional lethal gene knockout methods limit detailed functional studies. CRISPR interference (CRISPRi) technology enables knockdown of gene expression without eliminating gene function, facilitating the study of essential genes and their interactions with antibiotics [42].
Peptide Nucleic Acids as Antimicrobial Agents
PNAs are synthetic DNA mimics where the sugar-phosphate backbone is replaced by repeating N-2-aminoethyl glycine units, while the polyamide chain is covalently bonded to nucleobases [1]. This unique structure confers several advantages:
- Exceptional binding affinity and specificity for complementary DNA/RNA sequences
- Resistance to enzymatic degradation by nucleases and proteases
- Stability under extreme chemical and thermal conditions
- Neutral backbone that eliminates electrostatic repulsion with target nucleic acids
PNA oligomers can be synthesized by solid-phase synthesis similar to peptides, with the C-terminus attached to solid-phase resin and N-functionalities protected by appropriate protecting groups [1]. Recent advances in automated synthesis have significantly improved the efficiency and purity of PNA production [4] [5].
Automated Synthesis Platform for PPNAs
The automated high-purity fast-flow synthesis platform represents a transformative approach for PPNA manufacturing. This methodology utilizes the iChemAFS platform with high-temperature flow chemistry to accelerate production by nearly an order of magnitude compared to manual approaches [4] [27]. The system features a fully automated flow PNA synthesizer consisting of seven modules: central control computer, solution storage system, three HPLC pumps, three multiposition valves, heating elements, reaction zone, and UV-vis detector [5].
A breakthrough on-resin head-to-tail cyclization strategy for PNAs and PPNAs using a diaminonicotinic acid (DAN) linker enables precise control over chain elongation and cyclization, achieving 2- to 12-residue sequences with up to 95% crude purity [4] [27]. This approach supports extensive functionalization, including natural and non-natural amino acids, and enables late-stage modifications critical for optimizing antimicrobial activity.
Synthesis Workflow and Optimization
The following diagram illustrates the automated synthesis workflow:
Synthesis Optimization: Through in-line UV-vis monitoring combined with LC-MS and HPLC product characterization, researchers optimized PNA synthesis conditions. Key improvements included:
- High-temperature coupling (70°C) reducing each amide bond formation to 10 seconds
- Piperidine optimization for Fmoc-deprotection at elevated temperatures
- Prevention of piperidine adduct formation during deprotection steps
- Minimization of nucleobase adducts and deletion sequences through precise temperature control
This optimized protocol achieves remarkable efficiency, requiring only 3 minutes per complete synthesis cycle without needing capping or double couplings typically required in traditional synthesis [5].
Research Reagent Solutions
Table 1: Essential Research Reagents for PPNA Synthesis and Evaluation
| Reagent/Chemical | Function/Application | Specifications/Notes |
|---|---|---|
| Rink Amide Resin | Solid support for synthesis | 0.5 mmol/g loading capacity [5] |
| PNA Monomers | Building blocks for oligomer synthesis | Fmoc-protected, 10 equiv used per coupling [5] |
| PyAOP Activator | Peptide coupling reagent | 9.6 equiv used per coupling cycle [5] |
| DIEA Base | Base for coupling reactions | 30 equiv in DMF [5] |
| Piperidine | Fmoc deprotection | Optimal for high-temperature deprotection [5] |
| Diaminonicotinic Acid (DAN) Linker | On-resin cyclization | Enables head-to-tail cyclization [4] |
| Cell-Penetrating Peptides (CPPs) | Cellular delivery enhancement | Often (RXR)â‚„ sequence for bacterial penetration [43] |
| TFMSA/TFA | Cleavage from resin | Final cleavage to release product [1] |
Case Study: Targeting Essential Genes in Resistant Bacteria
Proof-of-Concept: NagZ Targeting in Pseudomonas aeruginosa
A compelling proof-of-concept study validated a PPNA targeting peptidoglycan recycling as a strategy to reduce AmpC β-lactamase hyperproduction and restore β-lactam efficacy in Pseudomonas aeruginosa [43]. The experimental workflow for this approach is illustrated below:
PPNA Design and Mechanism of Action
The NagZ-targeting PPNA was designed complementary to the PAO1 nagZ sequence (positions -9 to +3, with "A" from ATG codon defined as +1): [N→C termini: (RXR)₄XB-OCATGAAAAGTCC], where R represents arginine, X is 6-aminohexanoic acid, B is β-alanine, and -O- is ethylene glycol [43]. The (RXR)₄ sequence serves as the cell-penetrating peptide, while XB-O acts as a linker for conjugation with the PNA.
The antisense PPNA inhibits translation of the nagZ mRNA by hybridizing with its complementary sequence, causing mRNA degradation. NagZ (N-acetyl-β-D-glucosaminidase) is essential for peptidoglycan recycling and AmpC β-lactamase hyperproduction in P. aeruginosa. By targeting NagZ rather than AmpC directly, this approach circumvents potential compensatory resistance mechanisms while simultaneously attenuating virulence [43].
Quantitative Efficacy Data
Table 2: PPNA Efficacy Against Resistant P. aeruginosa Strains
| Strain/Parameter | PAdacBΔD (Mutant) | OFC2I4 (Clinical) | Notes/Methodology |
|---|---|---|---|
| nagZ mRNA Reduction | ~75% decrease | ~75% decrease | RT-PCR normalized to rpsL [43] |
| ampC mRNA Reduction | ~75% decrease | ~75% decrease | RT-PCR normalized to rpsL [43] |
| Ceftazidime MIC (alone) | 64 mg/L | 64 mg/L | Standard microdilution [43] |
| Ceftazidime MIC (+PPNA) | 8 mg/L | 8 mg/L | With 2 µM NagZ-PPNA [43] |
| PPNA MIC | 16 µM | 16 µM | Cytotoxicity limit [43] |
| Optimal PPNA Concentration | 2 µM | 2 µM | Balance of efficacy & safety [43] |
| Cytotoxicity | Low | Low | Human A549 cells [43] |
Experimental Protocols
PPNA Synthesis Protocol (Automated Flow Chemistry)
Materials: iChemAFS platform or equivalent automated synthesizer; Rink Amide resin (0.5 mmol/g); Fmoc-PNA monomers; PyAOP activator; DIEA base; Piperidine; DMF solvent; Cleavage cocktail (TFA/TFMSA)
Procedure:
- Resin Loading: Load 15 mg of Rink Amide resin (0.5 mmol/g) into the reactor body designed for 7.5 μmol scale synthesis.
- Monomer Preparation: Prepare PNA monomer solutions (10 equiv), activator PyAOP (9.6 equiv), and base DIEA (30 equiv) in DMF.
- Coupling Cycle: For each monomer addition:
- Merge monomer, activator, and base solutions through valves
- Heat mixture to 70°C to form activated ester
- Flow through resin bed maintained at 70°C (10 seconds per amide bond formation)
- Deprotection: Flow piperidine solution through room temperature loop to achieve ~40°C at reactor for efficient Fmoc removal.
- Cycle Repetition: Repeat steps 3-4 for sequence elongation (no capping or double couplings required).
- Cleavage: Cleave PPNA from resin using TFA/TFMSA cocktail.
- Purification: Purify crude product via RP-HPLC; characterize by MALDI MS [4] [5].
Bacterial Susceptibility Testing Protocol
Materials: Bacterial strains (e.g., P. aeruginosa PAdacBΔD, OFC2I4); Müller-Hinton broth; Non-binding U bottom-shaped 96-well plate; Gas-permeable sealing foils; NagZ-PPNA, Scr-PPNA (scrambled control), Ctrl-PPNA (random sequence control)
Procedure:
- Inoculum Preparation: Dilute bacterial strains to 5×10ⴠcells in non-adjusted Müller-Hinton broth.
- PPNA Treatment: Add decreasing concentrations of PPNAs (16 to 0.125 µM) to bacterial suspensions in 200 µL final volume.
- Incubation: Cover plates with gas-permeable sealing foils and incubate at 37°C with 180 rpm agitation for 14 hours.
- RNA Extraction: Harvest bacteria and purify RNA for real-time RT-PCR analysis.
- Gene Expression Analysis: Quantify nagZ and ampC expression using specific primers, normalizing to housekeeping gene rpsL.
- MIC Determination: Perform microdilution testing to determine MICs of ceftazidime alone and combined with PPNAs at fixed concentration (2 µM) [43].
Checkerboard Synergy Assay Protocol
Materials: Serial dilutions of β-lactam antibiotic; Fixed concentration of PPNA (2 µM); Bacterial inoculum as above; 96-well microtiter plates
Procedure:
- Antibiotic Preparation: Prepare two-fold serial dilutions of ceftazidime (or other β-lactam) in Müller-Hinton broth.
- PPNA Addition: Add fixed concentration of NagZ-PPNA (2 µM) to all antibiotic-containing wells.
- Inoculation: Add standardized bacterial inoculum (5×10ⴠCFU/well).
- Incubation: Incubate plates at 37°C for 18-24 hours.
- FIC Calculation: Determine Fractional Inhibitory Concentration (FIC) index using the formula: FIC index = (MIC of antibiotic in combination/MIC of antibiotic alone) + (MIC of PPNA in combination/MIC of PPNA alone). Synergy is defined as FIC index ≤0.5 [43].
Discussion and Future Perspectives
The integration of automated PPNA synthesis platforms with antisense strategies targeting essential bacterial genes represents a paradigm shift in antimicrobial development. The proof-of-concept study targeting nagZ in P. aeruginosa demonstrates that PPNA-mediated silencing of essential resistance genes can resensitize resistant strains to conventional antibiotics [43]. This approach offers several advantages:
- Sequence-specific targeting minimizes disruption of beneficial microbiota
- Modular design enables rapid adaptation to evolving resistance mechanisms
- Dual benefit of reducing resistance while potentially attenuating virulence
- Compatibility with existing antibiotics extends the lifespan of current therapeutics
Future research directions should focus on expanding the library of PPNAs targeting diverse essential genes identified through chemical genomics approaches [42], optimizing CPP sequences for enhanced species-specific delivery, and exploring combination therapies that target multiple resistance mechanisms simultaneously. The automated synthesis platform enables rapid production of PPNA variants for structure-activity relationship studies, accelerating the optimization of antimicrobial potency while minimizing cytotoxicity.
As antibiotic resistance continues to evolve, the integration of genomics-guided target identification with automated PPNA synthesis presents a promising pathway for developing precision antimicrobials that can address the growing threat of multidrug-resistant bacterial infections.
This application note details the quantitative frameworks and experimental protocols for validating the efficacy and safety of peptide-PNA conjugates (PPNAs) synthesized on automated platforms. The transition from traditional synthesis to automated, high-throughput flow chemistry has significantly accelerated the production of these promising antisense agents [5]. However, the full potential of this advanced manufacturing capability can only be realized with equally robust biological validation methods. This document provides standardized protocols for three critical assessment areas: splice-correction to confirm functional mRNA targeting, gene silencing to quantify antiviral or therapeutic activity, and cytotoxicity profiling to ensure therapeutic safety [5] [44]. These methods are specifically contextualized for evaluating PPNAs, which combine the exceptional binding affinity and nuclease resistance of PNAs with the improved cellular uptake provided by cell-penetrating peptides [5].
The following tables consolidate key quantitative benchmarks for assessing PPNA performance, integrating data from synthetic outcomes, functional efficacy, and safety profiles.
Table 1: Synthesis and Functional Efficacy Metrics for PPNAs
| Parameter | Representative Result | Experimental Context |
|---|---|---|
| Synthesis Crude Purity | Up to 95% | Automated flow synthesis of cyclic PNA/PPNA (2-12 residues) [4] |
| Synthesis Time Reduction | Order of magnitude faster | Automated flow vs. manual synthesis [5] |
| Splice-Correction Efficacy | Threefold activity increase vs. transfected PNA | EGFP assay with IVS2-654 targeting PPNA [5] |
| Antiviral Potency (SARS-CoV-2) | >95% viral titer reduction (IC~50~ = 0.8 μM) | PPNA targeting 5'UTR in live virus infection assay [5] |
| Splicing Correction Efficiency | Consistent, high correction in patient cells | EDSpliCE platform for deep intronic variants [45] |
Table 2: Cytotoxicity and Assay Performance Metrics
| Parameter | Method/Result | Significance/Context |
|---|---|---|
| Cytotoxicity (CC~50~) | Determined via MTS assay in uninfected cells [46] | Quantifies compound toxicity independent of antiviral effect. |
| Viability Assay (Barrier Integrity) | TEER & Lucifer Yellow flux [47] | Detects early cytotoxicity before cell death; more sensitive than colorimetric assays. |
| Selective Staining (Dead Cells) | Propidium Iodide, SYTOX Green [44] | Fluorescent dyes penetrate compromised membranes, enabling quantitative dead cell counting. |
| Therapeutic Index | CC~50~ / IC~50~ | Critical ratio defining window between safety and efficacy. |
Experimental Protocols
Splice-Correction Assay (EGFP System)
Principle: This functional assay quantifies the ability of a PPNA to correct a mutated β-globin intron (IVS2-654) inserted into an Enhanced Green Fluorescent Protein (EGFP) reporter gene. Successful splice-correction by an antisense PPNA restores proper EGFP splicing and translation, yielding measurable fluorescence [5].
Procedure:
- Cell Seeding: Seed HeLa cells stably expressing the EGFP-IVS2-654 reporter construct in a 96-well plate at a density of 2 x 10^4 cells/well and culture for 24 hours.
- PPNA Treatment: Transfert cells with the candidate PPNAs (e.g., 1-10 µM range) using a standard transfection reagent. Include controls:
- Negative Control: Untreated cells or scrambled PPNA sequence.
- Positive Control: Transfected with a known active antisense oligonucleotide (e.g., a commercial splice-corrector).
- Incubation: Incubate the cells for 24-48 hours at 37°C, 5% CO~2~ to allow for cellular uptake, splicing, and EGFP expression.
- Analysis:
- Flow Cytometry: Harvest cells, resuspend in PBS, and analyze using a flow cytometer (FITC channel). The percentage of EGFP-positive cells indicates successful splice-correction.
- Data Calculation: Normalize the mean fluorescence intensity (MFI) of treated samples to the positive control. Activity is often reported as a fold-increase over a negative control or a transfected PNA standard [5].
Live Virus Antiviral Assay (Gene Silencing)
Principle: This protocol assesses the efficacy of antisense PPNAs in inhibiting viral replication in a live virus infection model, as demonstrated for SARS-CoV-2 [5]. It is paired with a cytotoxicity assay to determine a selective index.
Procedure:
- Cell and Plate Preparation:
- Seed appropriate cells (e.g., Vero E6/TMPRSS2 for SARS-CoV-2) in two identical 96-well plates: one for the antiviral (infected) plate and one for the cytotoxicity (uninfected) plate [46].
- Culture cells until they reach 80-90% confluency.
- Compound Treatment:
- Prepare a serial dilution (e.g., 1:3, 8 points) of the PPNA candidates in infection media. The highest concentration row contains media without DMSO, while subsequent dilutions are made in media supplemented with DMSO to match the solvent concentration of the drug stock [46].
- Aspirate growth media from both plates and add 100 µL of the serial PPNA dilutions to the corresponding wells of both plates.
- Incubate plates for 2 hours at 37°C, 5% CO~2~ for prophylactic pretreatment.
- Virus Infection:
- Antiviral Plate: Infect cells with the target virus (e.g., SARS-CoV-2) at a predetermined Multiplicity of Infection (MOI) in a minimal volume of infection media.
- Cytotoxicity Plate: Perform a "mock infection" by adding 50 µL of plain 2% media to all wells [46].
- Incubate both plates for the duration of the virus infection cycle (e.g., 48-72 hours).
- Endpoint Analysis:
- Viral Titer Quantification: After the incubation period, quantify viral replication using a method such as plaque assay, TCID~50~, or RT-qPCR. The percentage reduction in viral titer is calculated relative to the virus-only control.
- Cytotoxicity Measurement: On the cytotoxicity plate, measure cell viability using an MTS or MTT assay per the manufacturer's instructions to determine the CC~50~ value [46].
Cytotoxicity Profiling Assays
Principle: Cytotoxicity must be evaluated to ensure that the biological activity of a PPNA is not due to general cell death. A multi-faceted approach is recommended, as traditional colorimetric assays may miss early toxic effects.
Table 3: Key Reagent Solutions for Cytotoxicity Assessment
| Reagent / Assay | Function / Principle | Key Considerations |
|---|---|---|
| MTS/MTT Assay | Measures metabolic activity as a proxy for viable cell number. | Can detect toxicity late, after significant damage [47]. |
| Propidium Iodide (PI) | Fluorescent DNA dye impermeable to live cells. Stains dead cells. | Ideal for endpoint assays; can be used in flow cytometry or plate readers [44]. |
| SYTOX Green | High-affinity nucleic acid stain impermeable to live cells. | >500-fold fluorescence enhancement upon DNA binding; suitable for HTS [44]. |
| Transepithelial Electrical Resistance (TEER) | Measures barrier integrity of cell monolayers. | Detects early cytotoxic events before cell death occurs [47]. |
| Lucifer Yellow (LY) Flux | Paracellular tracer to quantify barrier integrity. | Increased flux indicates compromised tight junctions, an early toxicity indicator [47]. |
A. Metabolic Inhibition (MTS) Assay for CC~50~ Determination
- Use the cytotoxicity plate from the antiviral assay protocol (Section 3.2).
- After the incubation period, add MTS reagent directly to each well.
- Incubate for 3-4 hours at 37°C to allow viable cells to convert MTS into a formazan product.
- Measure the absorbance at 490 nm (and optionally at 750 nm for background subtraction) using a plate reader [46].
- Calculate the percentage of cell viability relative to the untreated control wells. The CC~50~ is the concentration that reduces cell viability by 50%.
B. Early Cytotoxicity (Barrier Integrity) Assay
- Cell Culture: Seed Caco-2 or similar epithelial cells onto 96-well transwell inserts (e.g., CacoReady plates) and culture until a fully differentiated monolayer forms, confirmed by TEER values >500 Ω×cm² [47].
- PPNA Exposure: Expose the apical side of the monolayer to increasing concentrations of the PPNA for 24 hours.
- Measurement:
- TEER: Measure Transepithelial Electrical Resistance before and after exposure using a voltohmmeter. A decrease in TEER indicates a loss of barrier integrity.
- Lucifer Yellow Flux: After TEER measurement, add Lucifer Yellow to the apical chamber and incubate. Sample from the basolateral chamber and measure fluorescence (Ex/Em ~428/540 nm). An increase in flux percentage indicates compromised integrity [47].
- Stratification: Compounds can be stratified as non-toxic, moderately toxic, or very toxic based on the degree of change in TEER and LY flux compared to controls [47].
Research Reagent Solutions
Table 4: Essential Materials for PPNA Validation Studies
| Category | Item | Specific Example / Catalog Number | Function |
|---|---|---|---|
| Cell Lines | Reporter Cell Line | HeLa pEGFP-IVS2-654 [5] | Functional splice-correction assessment. |
| Virus-permissive Cells | Vero E6/TMPRSS2 [46] | Host for live virus antiviral assays. | |
| Assay Kits & Dyes | MTS Reagent | Sigma #11465007001 [46] | Colorimetric measurement of cell viability. |
| Dead Cell Stain | SYTOX Green (ThermoFisher S7020) [44] | Fluorescent, selective staining of dead cells for HTS. | |
| Dead Cell Stain | Propidium Iodide (ThermoFisher P3566) [44] | Classical DNA dye for dead cell identification. | |
| Specialized Plates | Barrier Integrity | CacoReady 96w plates [47] | Transwell inserts for TEER and flux measurements. |
| Critical Reagents | P-gp Inhibitor | e.g., 2 µM in infection media [46] | Optional; inhibits efflux pumps to prevent false negatives. |
Experimental and Data Analysis Workflows
The following diagrams illustrate the key experimental and analytical pathways described in this document.
PPNA Validation Workflow
Antiviral & Cytotoxicity Assay Setup
Within the field of peptide-peptide nucleic acid (PNA) conjugate research, the choice of synthesis methodology critically influences the success of downstream biological evaluation. The development of automated platforms for synthesizing these complex molecules has emerged as a pivotal innovation, with automated fast-flow synthesis presenting a compelling alternative to traditional batch methods. This application note provides a detailed comparative analysis of these two approaches, focusing on the critical parameters of yield, purity, and speed. The data and protocols herein are designed to equip researchers and drug development professionals with the empirical evidence needed to select the optimal synthesis strategy for their conjugate development programs, directly supporting advanced research within the context of an automated platform for peptide-PNA conjugate synthesis.
Comparative Performance Data
The quantitative comparison between automated fast-flow synthesis and traditional batch methods reveals significant advantages across multiple performance metrics, as summarized in the table below.
Table 1: Quantitative Comparison of Flow Synthesis vs. Traditional Batch Methods
| Performance Metric | Traditional Batch SPPS | Automated Fast-Flow SPPS | Experimental Context & Citation |
|---|---|---|---|
| Coupling Speed | 10 minutes per amide bond (at 45°C) [5] | a)10 seconds per amide bond [5] | PNA synthesis on a packed bed resin at 70°C [5]. |
| Overall Synthesis Time | Order of magnitude slower [27] | Reduced by nearly an order of magnitude [5] [27] | Synthesis of PNA and peptide-PNA conjugates (PPNAs) [5] [27]. |
| Crude Purity | ~57% (for a challenging 4-mer PNA) [5] | ~70% (for the same 4-mer PNA sequence) [5] | Direct comparison of the same PNA sequence (ACTG-Gly-CONH2) [5]. |
| Reagent Efficiency | Often requires large excesses (e.g., 6 equiv. monomer) [5] | Effective couplings with 1.2–10 equivalents of amino acid/PNA monomer [5] [48] | Enabled by precise reagent delivery and minimized reactor volume [5] [48]. |
| Solvent Consumption | High, with multiple wash steps between cycles [49] | ~70 mL per mmol per cycle; up to 95% waste reduction possible [48] [49] | Wash-free or reduced-wash protocols [48] [49]. |
| Scale-up | Requires re-optimization [48] | Direct scale-up from µmol to mmol scale without re-optimization [48] | Syntheses optimized at 50 µmol scaled directly to 30 mmol [48]. |
a) This rate includes the coupling reaction only, within a total cycle time of approximately 3 minutes [5].
Experimental Protocols
Protocol 1: Automated Fast-Flow Synthesis of PNA Conjugates
This protocol is adapted from the methodology used to synthesize anti-SARS-CoV-2 PPNAs on a fully automated fast-flow instrument [5].
Objective: To synthesize a peptide-PNA conjugate (PPNA) using an automated fast-flow synthesizer. Key Advantages: High speed, high crude purity, and minimal reagent use.
Materials and Reagents:
- Resin: Rink Amide resin (0.5 mmol/g loading) [5].
- Monomers: Fmoc/Bhoc-protected PNA monomers and Fmoc-protected amino acids [5] [29].
- Activator: PyAOP ( (7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate) [5].
- Base: N,N-Diisopropylethylamine (DIEA) [5].
- Solvent: Anhydrous DMF [5].
- Deprotection Reagent: 20% Piperidine in DMF [5].
Procedure:
- Reactor Setup: Pack 15 mg of Rink Amide resin (for a ~7.5 μmol scale synthesis) into the flow reactor's packed bed [5].
- Monomer Activation: For each coupling cycle, the system automatically draws reagents from storage. The PNA monomer (10 equiv), activator (PyAOP, 9.6 equiv), and base (DIEA, 30 equiv) are merged and passed through a heating module at 70°C to form the activated ester [5].
- Coupling Reaction: The activated monomer solution flows through the resin-packed reactor, which is also maintained at 70°C. The residence time for amide bond formation is 10 seconds [5].
- Fmoc Deprotection: A solution of 20% piperidine in DMF flows through the system to remove the Fmoc protecting group. The system is configured to generate a ~40°C environment for efficient deprotection with minimized side reactions [5].
- Cycle Repetition: Steps 2-4 are repeated automatically until the desired PNA sequence is complete. The CPP peptide can be synthesized directly onto the PNA sequence in a "single-shot" manner without intermediate cleavage [5].
- Cleavage and Isolation: Upon completion, the conjugate is cleaved from the resin using a standard cleavage cocktail (e.g., TFA:Water:TIPS, 95:2.5:2.5). The crude product is precipitated and washed with cold diethyl ether before analysis and purification [5] [29].
Monitoring: The flow synthesizer used in the cited research featured an in-line UV-vis detector to monitor the Fmoc-removal chromatogram in real-time, allowing for immediate detection of coupling or deprotection inefficiencies [5].
Protocol 2: Traditional Batch Synthesis of PNA Conjugates
This protocol outlines a standard Fmoc-based batch synthesis for PNA, which is often limited to shorter sequences or requires capping and double-coupling strategies for longer ones [5] [33].
Objective: To synthesize a PNA oligomer using traditional batch solid-phase synthesis. Key Challenges: Longer cycle times, lower crude purity for challenging sequences, and higher reagent consumption.
Materials and Reagents:
- Resin: Rink Amide resin or Fmoc-XAL-PEG-PS resin [7] [29].
- Monomers: Fmoc/Bhoc-protected PNA monomers [7] [29].
- Activator: HCTU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate) or similar carbodiimide-based reagents [29].
- Base: DIPEA (N,N-Diisopropylethylamine) [29].
- Solvent: DMF or NMP (N-Methyl-2-pyrrolidone) [29].
- Deprotection Reagent: 20% Piperidine in DMF [29].
Procedure:
- Resin Swelling: Place the resin (e.g., 100 μmol scale) in a reaction vessel and swell with DCM or DMF for 20-30 minutes [29].
- Fmoc Deprotection: Drain the solvent and treat the resin with 20% piperidine in DMF (2 x 3-5 minutes) at room temperature to remove the Fmoc group [29].
- Washing: Drain the deprotection solution and wash the resin 5 times with DMF to remove residual piperidine completely [49].
- Coupling Reaction: Add a solution of the Fmoc-PNA monomer (3-6 equiv) and activator (e.g., HCTU, 3 equiv) in DMF/NMP, followed by base (e.g., DIPEA, 6 equiv). Allow the coupling to proceed for ~30-60 minutes at room temperature with agitation [5] [29].
- Washing: Drain the coupling solution and wash the resin 3-5 times with DMF [49].
- Cycle Repetition: Repeat steps 2-5 for each additional monomer in the sequence.
- Final Cleavage: After the final deprotection, cleave the PNA from the resin using a TFA-based cocktail (e.g., TFA:Water:TIPS, 95:2.5:2.5) for 1.5-3 hours. Precipitate the crude product in cold diethyl ether, isolate by centrifugation, and purify by RP-HPLC [29].
Workflow and System Visualization
The fundamental difference between the two methodologies lies in the reactor design and reagent delivery, which directly impacts efficiency and outcomes.
Diagram 1: A comparison of the fundamental workflows for fast-flow and traditional batch synthesis, highlighting key operational differences.
The Scientist's Toolkit: Research Reagent Solutions
The synthesis of peptide-PNA conjugates requires specialized reagents and solid supports to achieve successful results.
Table 2: Essential Materials for Peptide-PNA Conjugate Synthesis
| Reagent / Material | Function / Role | Examples & Notes |
|---|---|---|
| PNA Monomers | Building blocks for PNA chain elongation. | Fmoc-A(Bhoc)-OH, Fmoc-G(Bhoc)-OH, etc. Bhoc protects the nucleobase amino groups [5] [7]. |
| Solid Support | Platform for solid-phase synthesis. | Rink Amide Resin (for C-terminal amide), Fmoc-XAL-PEG-PS, TentaGel. Low-loading, hydrophilic resins help reduce aggregation [33] [7] [29]. |
| Coupling Activators | Activate carboxylic acid for amide bond formation. | PyAOP, HCTU. Chosen for efficient PNA monomer coupling and compatibility with heated flow chemistry [5] [29]. |
| Deprotection Base | Removes the temporary Fmoc protecting group. | 20% Piperidine in DMF. In wash-free batch, pyrrolidine is sometimes used for easier evaporation [5] [49]. |
| Cleavage Cocktail | Cleaves final product from resin and removes permanent protecting groups. | TFA:Water:TIPS (95:2.5:2.5). Scavengers like TIPS and water prevent side reactions during cleavage [5] [29]. |
| Spacer | Links functional modules (e.g., PNA to peptide). | 8-Amino-3,6-dioxaoctanoic acid (PEG spacer). Introduces flexibility and reduces steric hindrance between domains [29]. |
The empirical data and protocols presented demonstrate that automated fast-flow synthesis represents a significant technological advancement over traditional batch methods for the production of peptide-PNA conjugates. The flow platform offers a compelling combination of dramatically increased speed, enhanced crude purity, superior reagent efficiency, and straightforward, reproducible scale-up. For research teams focused on rapidly generating and screening libraries of peptide-PNA conjugates for drug discovery, the adoption of an automated fast-flow synthesis platform can significantly accelerate the iterative cycle of design, synthesis, and biological testing, thereby streamlining the entire development pipeline.
Conclusion
Automated fast-flow synthesis has emerged as a robust and enabling platform that directly addresses the historical bottlenecks in peptide-PNA conjugate development. By drastically accelerating synthesis times, improving crude purity, and enabling the straightforward production of complex conjugates, this technology is poised to accelerate the pre-clinical pipeline. The validated success of PPNAs as potent antisense agents against high-priority targets like SARS-CoV-2 and multidrug-resistant bacteria underscores their significant therapeutic potential. Future directions will likely focus on expanding the scope of deliverable cargoes, refining CPP specificity to enhance tissue targeting, and advancing these promising conjugates toward clinical translation, ultimately opening new frontiers in precision medicine and antisense therapy.
References
- 1. Peptide Nucleic Acids: Recent Developments in the ... [https://www.sciencedirect.com/science/article/abs/pii/S0045206823005217]
- 2. Insights on chiral, backbone modified peptide nucleic acids [https://pmc.ncbi.nlm.nih.gov/articles/PMC5329900/]
- 3. A Bright Combination for a FIT-PNA RNA Sensor [https://www.frontierspartnerships.org/journals/british-journal-of-biomedical-science/articles/10.3389/bjbs.2025.15526/full]
- 4. Automated high-purity fast-flow synthesis of cyclic peptide- ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725076752]
- 5. Automated Flow Synthesis of Peptide–PNA Conjugates - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8874765/]
- 6. Automated high-purity fast-flow synthesis of cyclic peptide- ... [https://research.mpu.edu.mo/en/publications/automated-high-purity-fast-flow-synthesis-of-cyclic-peptide-pna-c/]
- 7. Application of Cell-Penetrating Peptides (CPP) to Enhance ... [https://link.springer.com/article/10.1007/s10989-025-10776-1]
- 8. Chemical approaches to discover the full potential of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8313981/]
- 9. Peptide nucleic acid conjugates and their antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10618302/]
- 10. The challenge of peptide nucleic acid synthesis - Chemical Society... [https://pubs.rsc.org/en/content/articlehtml/2023/cs/d2cs00049k]
- 11. The Flexibility of PNA Synthesis [https://www.biosearchtech.com/support/nac/flexibility-of-pna-synthesis]
- 12. Peptide Nucleic Acid (PNA) Synthesis [https://www.biosearchtech.com/support/nac/peptide-nucleic-acid-pna-synthesis]
- 13. Cell-penetrating peptide - Wikipedia [https://en.wikipedia.org/wiki/Cell-penetrating_peptide]
- 14. Cell-penetrating peptides: tools for intracellular delivery of ... [https://pubmed.ncbi.nlm.nih.gov/15968462/]
- 15. Cell-Penetrating Peptides (CPPs) as Therapeutic and Diagnostic Agents for Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9688740/]
- 16. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4034016/]
- 17. Universal peptide synthesis via solid-phase methods fused ... [https://www.nature.com/articles/s41467-025-62344-2]
- 18. Automated Peptide Synthesis [https://americanpeptidesociety.org/explore/automated-peptide-synthesis/]
- 19. Getting Started with Automated Peptide Synthesis Guide [https://www.gyrosproteintechnologies.com/purepep-blog/getting-started-with-automated-peptide-synthesis-guide-purepep-blog]
- 20. Synthetic developments towards PNA–peptide conjugates [https://www.sciencedirect.com/science/article/abs/pii/S1367593103001418]
- 21. Fully automated fast-flow synthesis of antisense ... [https://www.nature.com/articles/s41467-021-24598-4]
- 22. Rapid Flow-Based Peptide Synthesis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4045704/]
- 23. Automated Flow Synthesis of Peptide–PNA Conjugates [https://pentelutelabmit.com/portfolio_page/automated-flow-synthesis-of-peptide-pna-conjugates/]
- 24. How Do You Reconstitute Peptide Nucleic Acids (PNAs) [https://www.biosyn.com/faq/How-Do-You-Reconstitute-Peptide-Nucleic-Acids-(PNAs).aspx]
- 25. Evaluation of Peptide Nucleic Acid Encapsulation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606638/]
- 26. Antiviral Peptide-Based Conjugates: State of the Art and ... [https://www.mdpi.com/1999-4923/15/2/357]
- 27. Automated high-purity fast-flow synthesis of cyclic peptide- ... [https://ui.adsabs.harvard.edu/abs/2025ChEnJ.52166837Z/abstract]
- 28. Automated Fast-Flow Synthesis of Peptide Nucleic Acid ... [https://chemrxiv.org/engage/chemrxiv/article-details/60c75562f96a004682288863]
- 29. An Efficient Approach for the Design and Synthesis ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.843163/full]
- 30. Blog - Cyanobenzothiazole (CBT) Derivatives for “Click†... [https://iris-biotech.de/global/blog/cyanobenzothiazole-cbt-derivatives-for-click-conjugation.html]
- 31. Recent applications of CBT-Cys click reaction in biological ... [https://www.sciencedirect.com/science/article/abs/pii/S0968089622002735]
- 32. A Template-Mediated Click-Click Reaction: PNA-DNA, PNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3597112/]
- 33. Peptide nucleic acids (PNA) – Overview [https://intavispeptides.com/de/insights/25]
- 34. Recent Advances in Chemical Modification of Peptide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3443988/]
- 35. High Quality Custom PNA Oligo Synthesis Service [https://pnabio.com/custom-pna-oligos/]
- 36. An Efficient Approach for the Design and Synthesis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8964499/]
- 37. Multipep 2 - Automated Parallel Peptide Synthesizer [https://cem.com/multipep2]
- 38. Unveiling the promise of peptide nucleic acids as ... [https://pubs.rsc.org/en/content/articlelanding/2025/cb/d4cb00274a]
- 39. Late-Stage Serine Modification Enables Noncanonical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12426918/]
- 40. Cell-penetrating peptide conjugates of peptide nucleic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1301599/]
- 41. Genomics for antimicrobial resistance—progress and future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12057382/]
- 42. Chemical genomics informs antibiotic and essential gene ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1011642]
- 43. An antisense peptide-conjugated peptide nucleic acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403870/]
- 44. Cytotoxicity Assays: In Vitro Methods to Measure Dead Cells [https://www.ncbi.nlm.nih.gov/books/NBK540958/]
- 45. New Techniques to Fix Splicing Errors Seen in Genetic Disorders [https://scisimple.com/en/articles/2025-11-03-new-techniques-to-fix-splicing-errors-seen-in-genetic-disorders--ak2dryn]
- 46. Cytotoxicity Screening Assay - Paired with Antiviral Assays [https://www.protocols.io/view/cytotoxicity-screening-assay-paired-with-antiviral-diky4cxw.html]
- 47. Cytotoxicity Assay Protocol [https://medtechbcn.com/readycell/blog/cytotoxicity-assay-protocol/]
- 48. Learn Why Flow Outperforms Batch for Peptide Synthesis [https://www.vapourtec.com/blog/flow-vs-batch-solid-phase-peptide-synthesis/]
- 49. Total wash elimination for solid phase peptide synthesis [https://www.nature.com/articles/s41467-023-44074-5]