Frontside Attack in Nucleophilic Substitution: Unraveling the Stereochemical Forbidden Pathway and its Implications for Reaction Design
This article provides a comprehensive analysis of the frontside attack mechanism in nucleophilic substitution reactions, a pathway stereochemically forbidden in classic SN2 processes.
Frontside Attack in Nucleophilic Substitution: Unraveling the Stereochemical Forbidden Pathway and its Implications for Reaction Design
Abstract
This article provides a comprehensive analysis of the frontside attack mechanism in nucleophilic substitution reactions, a pathway stereochemically forbidden in classic SN2 processes. Tailored for researchers and drug development professionals, we explore the fundamental principles governing reaction stereochemistry, including the orbital interactions that enforce backside displacement and result in inversion of configuration. The content extends to methodological approaches for studying reaction dynamics, troubleshooting factors that potentially enable frontside-like pathways, and comparative validation against established SN2 and SN1 mechanisms. Special emphasis is placed on the implications for pharmaceutical synthesis where precise stereochemical control is paramount, incorporating recent advances in understanding reaction pathways beyond the traditional SN2 paradigm.
Stereochemical Fundamentals: Why Frontside Attack is Stereochemically Forbidden in SN2 Reactions
Defining Frontside vs. Backside Attack Pathways in Nucleophilic Substitution
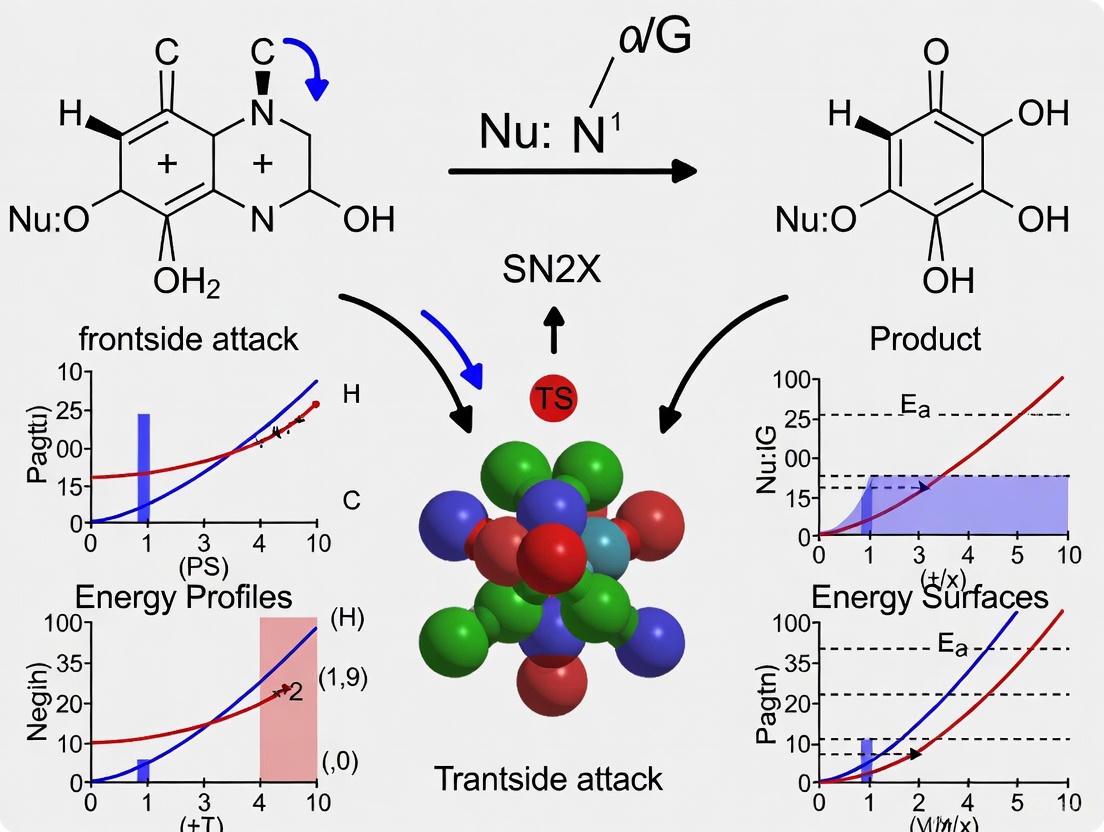
In nucleophilic substitution reactions, the spatial trajectory of the attacking nucleophile relative to the departing leaving group fundamentally dictates the stereochemical outcome and reaction mechanism. Two distinct pathways are defined: the backside attack, where the nucleophile approaches the electrophilic carbon from the side opposite the leaving group, and the frontside attack, where approach occurs from the same side as the leaving group [1]. The backside attack is the hallmark of the concerted, bimolecular SN2 mechanism, resulting in a single-step process with inversion of stereochemical configuration at the carbon center [2] [3]. In stark contrast, a frontside attack would theoretically lead to retention of configuration but is sterically and electronically disfavored in typical SN2 reactions due to repulsion between the incoming nucleophile and the departing leaving group [1] [2]. This technical guide frames this core stereochemical concept within emerging research on alternative mechanisms, specifically the Halogenophilic Nucleophilic Substitution (SN2X) reaction, where distinct pathways, potentially involving frontside-associated complexes, can compete with or complement the classic SN2 route [4] [5].
Mechanistic Foundations: SN2, SN1, and the Emergence of SN2X
The SN2 Mechanism and Backside Attack
The SN2 mechanism is characterized by a concerted, single-step process in which bond formation between the nucleophile and the electrophilic carbon is synchronous with bond cleavage to the leaving group. The kinetics are second-order, dependent on both nucleophile and substrate concentration [3] [6]. The requirement for backside attack is absolute in standard organic solvents; the nucleophile must approach 180° opposite the C–LG bond to effectively overlap with the σ* antibonding orbital of that bond, facilitating its rupture [3] [6]. This trajectory results in a trigonal bipyramidal transition state and inevitable inversion of configuration (Walden inversion) at chiral centers [2] [3]. Steric hindrance around the electrophilic carbon (e.g., tertiary centers) dramatically slows or prevents this approach, favoring alternative mechanisms like SN1 [6].
The SN1 Mechanism and Loss of Stereospecificity
The unimolecular SN1 mechanism proceeds via a stepwise process. The rate-determining step is the heterolytic cleavage of the C–LG bond to form a planar, sp²-hybridized carbocation intermediate [2] [7]. This intermediate can then be attacked by a nucleophile from either face. While this often leads to racemization for chiral substrates, complete racemization is not always observed because the departing leaving group can shield one face (the frontside), leading to a preference for backside attack on the carbocation and a net partial inversion [7].
The SN2X Mechanism: A Competing Pathway
Recent research has quantitatively characterized the Halogenophilic Nucleophilic Substitution (SN2X) pathway [4] [5]. This mechanism is distinct from SN2 but can yield identical products. It is proposed to proceed through a frontside attack-associated complex or a halogenophilic interaction where the nucleophile initially interacts with the halogen atom of the leaving group rather than the carbon center [4]. This pathway involves a pro-chiral anion intermediate, contrasting with the stereospecific, single-step SN2 mechanism. The coexistence of SN2 and SN2X pathways in reactions highlights a continuum of nucleophilic substitution mechanisms rather than strictly discrete categories [4].
The following tables consolidate key thermodynamic and kinetic parameters from seminal studies on attack pathways and the SN2X mechanism.
Table 1: Experimental Thermodynamic Data for Ion-Molecule Complex Formation via Frontside Attack (Gas Phase) [8]
| Reaction Complex | ΔH (kcal mol⁻¹) | ΔS (cal mol⁻¹ K⁻¹) |
|---|---|---|
| Cl⁻(BrCF₃) | -16.5 ± 0.2 | -24.5 ± 1 |
| Cl⁻(ICF₃) | -23.6 ± 0.2 | Not Reported |
| Br⁻(BrCF₃) | -13.9 ± 0.2 | -22.2 ± 1 |
Note: This data, obtained via pulsed-ionization high-pressure mass spectrometry, demonstrates the stability of frontside attack complexes in the gas phase, which precede the lower-energy backside attack SN2 transition state [8].
Table 2: Key Quantitative Parameters in SN2X Reaction Analysis [4] [5]
| Parameter | Symbol | Description |
|---|---|---|
| Halogenophilic Percentage | X% | The fraction of the product formed via the SN2X pathway in a reaction where both SN2 and SN2X are possible. Measured via kinetic simulations. |
| Relative Halogenophilicity | H | A quantitative parameter describing the intrinsic tendency of a system to undergo the SN2X pathway. Correlates with Hammett and Mayr parameters. |
| Overall Reaction Rate Constant | k | Governed by the contributions of both pathways: k = kₛₙ₂ + kₛₙ₂ₓ. |
Detailed Experimental Protocols
Objective: To map the potential energy surfaces for SN2 reactions between halide ions (X⁻) and trifluoromethyl halides (CF₃Y) and identify frontside vs. backside attack mechanisms.
Methodology:
- Computational Analysis:
- Level of Theory: Density Functional Theory (DFT) computations at the B3LYP/6-311+G(3df)//B3LYP/6-311+G(d) level.
- Procedure: Optimize geometries of all reactants, possible ion-molecule complexes, and transition states. Calculate vibrational frequencies to confirm stationary points (minima or first-order saddle points). Perform intrinsic reaction coordinate (IRC) calculations to connect transition states to corresponding minima. Calculate single-point energies to construct potential energy surfaces.
- Experimental Validation via Pulsed-Ionization High-Pressure Mass Spectrometry:
- Apparatus: A high-pressure mass spectrometer equipped with a pulsed ionization source.
- Procedure: Introduce a mixture of a halide ion precursor (e.g., CH₃Cl for Cl⁻) and the neutral substrate (CF₃Y) at a controlled pressure (typically 1-10 Torr) into the reaction chamber.
- Ionization: Generate thermalized reactant ions (X⁻) via a short pulse of electrons or photons.
- Reaction & Detection: Allow ions to react with neutrals for a controlled time (milliseconds to seconds). Mass-analyze and detect the resulting ions.
- Data Analysis: Measure the equilibrium concentrations of the reactant ions, product ions (Y⁻), and any observable ion-molecule complexes (X⁻(CF₃Y)). From the temperature dependence of the equilibrium constants, derive the enthalpy (ΔH) and entropy (ΔS) changes for complex formation using van't Hoff plots.
Objective: To determine the fraction of product formed via the SN2X pathway in a stereoselective reaction.
Methodology:
- Substrate Design: Employ a pro-chiral or chiral substrate where the SN2 pathway is stereospecific (leading to inversion), while the SN2X pathway proceeds via a pro-chiral anion intermediate that can lead to a different stereochemical outcome or racemization.
- Kinetic Experiment: Conduct the nucleophilic substitution reaction under standardized conditions. Periodically sample the reaction mixture.
- Stereochemical Analysis: Use chiral analytical techniques (e.g., Chiral HPLC or NMR spectroscopy with chiral shift reagents) to determine the enantiomeric excess (ee) or diastereomeric ratio of the product over time.
- Kinetic Simulation: Develop a kinetic model incorporating rate constants for both the SN2 (kₛₙ₂) and SN2X (kₛₙ₂ₓ) pathways, as well as the epimerization of any intermediate. Fit the experimental time-course data for product stereoisomer ratios to this model using simulation software.
- Calculation of X%: From the fitted rate constants, calculate the halogenophilic percentage: X% = [kₛₙ₂ₓ / (kₛₙ₂ + kₛₙ₂ₓ)] × 100%.
Visualization of Pathways and Workflows
Diagram 1: Competing Frontside and Backside Attack Pathways.
Diagram 2: Experimental Workflow for Quantifying SN2X Contribution.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Investigating Attack Pathways and SN2X Mechanisms
| Reagent / Material | Function in Research | Key Reference |
|---|---|---|
| Trifluoromethyl Halides (CF₃X, X=Cl, Br, I) | Model electrophilic substrates for gas-phase studies. The strong C–F bonds minimize complications from competing elimination, allowing clear study of substitution pathways. | [8] |
| Tetraalkylammonium Halide Salts (e.g., [ⁿBu₄N]⁺X⁻) | Source of "naked" or poorly solvated halide nucleophiles (X⁻) in organic solvents, maximizing nucleophilicity for studying SN2 kinetics and competition with SN2X. | Implied in [6] |
| Chiral or Pro-Chiral Alkyl Halides | Substrates designed with stereogenic centers or pro-chiral centers to track stereochemical fate, enabling discrimination between stereospecific SN2 and other pathways like SN2X. | [4] [5] |
| Polar Aprotic Solvents (DMSO, DMF, MeCN) | Solvents that dissolve ionic reagents while minimally solvating anions, thereby enhancing nucleophile reactivity for SN2 reactions. Used in kinetic studies of pathway competition. | [6] |
| Chiral Derivatizing Agents & HPLC Columns | Essential for stereochemical analysis of reaction products. Used to determine enantiomeric excess and track stereochemistry over time in kinetic experiments for X% calculation. | [4] [5] |
| Computational Software (Gaussian, ORCA, etc.) with DFT Functionals (e.g., B3LYP) | For mapping potential energy surfaces, identifying transition states (backside vs. frontside), and calculating thermodynamic parameters (ΔH, ΔS) to support experimental data. | [8] [9] |
Orbital Symmetry and Stereoelectronic Requirements for Effective Bond Formation
This technical guide examines the orbital symmetry and stereoelectronic prerequisites for effective bond formation, with a specific focus on the frontside attack pathway in nucleophilic substitution (SN2) reactions. Framed within broader thesis research on the SN2X (X denoting frontside attack) mechanism, we synthesize current experimental and computational evidence to delineate the precise geometric and electronic conditions necessary for bond-making and bond-breaking events. The analysis underscores that deviation from the classic backside attack paradigm introduces unique stereoelectronic constraints, which are quantified through kinetic studies, isotope effects, and trajectory dynamics. This resource is designed for researchers and drug development professionals seeking to understand or manipulate reaction pathways where stereoelectronics govern selectivity and outcome.
The bimolecular nucleophilic substitution (SN2) reaction is a cornerstone of mechanistic organic chemistry, traditionally characterized by a concerted backside attack leading to Walden inversion [9]. However, the potential energy surface (PES) for these reactions is rich with alternative pathways, including the frontside attack retention mechanism and the more recently discovered double-inversion pathway [10]. The feasibility of these non-canonical routes is governed not by thermodynamics alone, but by stringent orbital symmetry and stereoelectronic requirements. Effective bond formation during nucleophilic attack demands a specific spatial alignment of the nucleophile's donor orbital with the acceptor orbital (typically the σ* orbital of the carbon-leaving group bond) on the substrate. This guide details these requirements, placing the frontside attack (SN2X) mechanism within a coherent theoretical and experimental framework.
Theoretical Foundations: Orbital Interactions and Stereoelectronics
Orbital Symmetry in Backside vs. Frontside Attack
The classic SN2 mechanism proceeds via a backside attack where the nucleophile's highest occupied molecular orbital (HOMO) interacts with the substrate's lowest unoccupied molecular orbital (LUMO), which is the antibonding (σ*) orbital of the C–LG (leaving group) bond. This interaction is symmetry-allowed and maximized in an antiperiplanar approach, leading to a linear transition state (X–C–LG ~ 180°) and inversion of configuration.
In a frontside attack, the nucleophile approaches from the same side as the leaving group. This geometry forces an interaction between the nucleophile's HOMO and a different acceptor orbital on the substrate. Computational studies indicate that this pathway often involves a transition state with a much narrower X–C–LG angle (typically 40–100°) and is symmetry-disallowed under standard frontier molecular orbital theory for a direct displacement, resulting in a higher energy barrier [10]. The double-inversion mechanism, a distinct retention pathway, circumvents this by proceeding through a proton-abstraction step followed by a classic inversion, demonstrating how stereoelectronic constraints can be bypassed via a multi-step process [10].
Stereoelectronic Effects in Elimination and Substitution
Stereoelectronic principles are paramount in differentiating between competing E2 and SN2 pathways. For an E2 elimination to proceed concertedly, the proton being abstracted and the leaving group must be antiperiplanar to allow for optimal overlap in the transition state for π-bond formation [11]. This requirement is a quintessential stereoelectronic effect. Similarly, the preference for certain conformations in SN2 reactions, even for frontside pathways, can be traced to the need for orbital alignment. In cyclic systems, such as the dehydrohalogenation of substituted cyclohexanes, the antiperiplanar arrangement is a prerequisite for the E2 reaction, highlighting how molecular rigidity enforces stereoelectronic control [11].
Quantitative Data and Energetic Landscapes
The following tables summarize key quantitative data underpinning the stereoelectronic analysis of SN2 pathways.
Table 1: Deuterium Kinetic Isotope Effects (KIE) for Mechanism Discrimination
| Reaction System | kH/kD Ratio | Inferred Mechanism | Implication for Stereoelectronics | Source Context |
|---|---|---|---|---|
| 1-Bromo-2-phenylethane vs. dideuterated | 7.1 | E2 | C–H/D bond broken in rate-determining step; requires precise antiperiplanar alignment. | [11] |
| Theoretical for E1 mechanism | ~1 (No effect) | E1 | Proton loss occurs after rate-determining step; no strict stereoelectronic requirement in this step. | [11] |
Table 2: Computational Energy Barriers and Trajectory Analysis for SN2@C Reactions
| Reaction System | Central Barrier (ΔE≠,centr) | Overall Barrier (ΔE≠,ovr) | Dominant Retention Pathway at Low Collision Energy | TS Attack Angle (Frontside) | TS Attack Angle (Double Inversion) |
|---|---|---|---|---|---|
| F− + CH3Cl (Gas Phase) | Positive | Can be negative | Double Inversion | 40–100° | 120–180° (2nd step) |
| F− + CH3I (Gas Phase) | Lower than for CH3Cl | - | Mixed (Double Inversion dominant at low E) | 40–100° | 120–180° (2nd step) |
| Cl− + CH3Cl (Archetypal) | Positive | - | Frontside attack (high barrier) | - | - |
Note: Data synthesized from computational dynamics studies [10] [9]. The "attack angle" is defined as the ∠(X–C–LG) in the transition state region.
Table 3: Influence of Base and Substituents on Regiochemistry and Pathway Competition
| Variable | Effect on SN2 (E2) Pathway | Stereoelectronic Rationale |
|---|---|---|
| Base Strength (e.g., OH− vs. ROH) | Strong base promotes E2 over SN2 under basic conditions [9]. | Stronger base more effectively abstracts proton in antiperiplanar arrangement for E2. |
| Base Steric Hindrance (e.g., t-BuOK) | Increases proportion of least substituted alkene (non-Zaitsev) in E2; increases E2:SN2 ratio [11]. | Hindered base abstracts less sterically hindered proton, overriding thermodynamic stability of alkene product. |
| α- and β-Substituents on Substrate | Profoundly affects SN2 and E2 barriers (e.g., allyl, benzyl groups lower barriers) [9]. | Changes in substrate LUMO energy and geometry alter orbital overlap efficiency with nucleophile or base. |
| Solvent Polarity | Ionic SN2: Barrier increases (double-well to unimodal PES). Menshutkin (neutral): Barrier decreases [9]. | Solvation stabilizes localized charge (reactants) over delocalized charge (TS), or stabilizes charge separation. |
Experimental Protocols for Investigating Stereoelectronic Requirements
Protocol A: Measuring Primary Deuterium Kinetic Isotope Effects (KIE)
Objective: To determine if a C–H bond is broken in the rate-determining step, distinguishing between concerted (E2/SN2) and stepwise (E1/SN1) mechanisms with stereoelectronic consequences. Methodology:
- Synthesis: Prepare the natural abundance substrate (e.g., 1-bromo-2-phenylethane) and its specifically deuterated analogue (e.g., 1-bromo-2,2-dideuterio-2-phenylethane) using established synthetic routes (e.g., reduction of a ketone precursor with LiAlD4) [11].
- Kinetic Measurements: Under identical, controlled conditions (temperature, solvent, base concentration), subject both substrates to the reaction (e.g., dehydrohalogenation with a strong base like ethoxide).
- Rate Determination: Monitor the reaction progress using a suitable technique (e.g., gas chromatography, NMR spectroscopy) to determine the rate constants (kH and kD) for the consumption of starting material or formation of product.
- KIE Calculation & Interpretation: Calculate the ratio kH/kD. A significant primary KIE (typically >2, value of 7.1 observed [11]) indicates C–H bond cleavage in the rate-determining step, supporting a concerted E2 mechanism with an antiperiplanar requirement. A negligible KIE (~1) suggests C–H bond breaking occurs after the rate-determining step, consistent with an E1 mechanism where no specific stereoelectronic alignment is needed in the initial step.
Protocol B: Trajectory Analysis for SN2 Frontside vs. Double-Inversion Pathways
Objective: To numerically separate and identify frontside attack and double-inversion retention trajectories in gas-phase SN2 reaction dynamics. Methodology:
- Potential Energy Surface (PES) Generation: Develop a high-level analytical or ab initio PES for the reaction system (e.g., F− + CH3Cl) [10].
- Quasi-Classical Trajectory Calculations: Run millions of trajectories by sampling initial conditions (collision energy, impact parameter, internal rotations/vibrations) relevant to experimental crossed-beam setups.
- Vector-Projection Analysis (Configuration Determination):
- For each reactive trajectory, define vectors v1 (C to a substituent, e.g., H) and v2 (C to the leaving group, LG) in the reactant.
- In the product, define corresponding vectors v1' (C to the same substituent) and v2' (C to the nucleophile, Nu).
- Compute the scalar triple product S = v1 · (v2 × v1'). A negative sign of S indicates inversion of configuration; a positive sign indicates retention [10].
- Pathway Separation via TS Attack Angle:
- For retention trajectories, analyze the approach angle. Follow the leaving group's position backward from the products to the transition state region.
- Calculate the attack angle θ = ∠(Nu–C–LG) in this TS region.
- Classify: Trajectories with θ between 40° and 100° are assigned to the frontside attack pathway. Trajectories where this angle is between 120° and 180° (characteristic of a Walden-inversion TS, which is part of the double-inversion mechanism) are assigned to the double-inversion pathway [10].
- Validation: Cross-check assignments by animating subsets of trajectories to visually confirm the mechanism (direct frontside attack vs. proton-abstraction-then-inversion).
Protocol C: Microsolvation Studies on SN2 Reactivity
Objective: To probe the gradual effect of solvation on SN2 PES shape and mechanism, affecting charge localization and stereoelectronic demands. Methodology:
- Cluster Formation: Generate microsolvated nucleophile clusters (e.g., X−(H2O)_n, n=1-6) in the gas phase using supersonic expansion or ion-mobility techniques [9].
- Reactivity Measurements: Use guided ion beam mass spectrometry or flowing afterglow techniques to measure reaction rate constants as a function of cluster size (n) for reactions like Cl−(H2O)_n + CH3Br.
- Computational Modeling: Perform ab initio calculations to locate minima (reactant/ion-dipole complexes) and transition states on the PES for different n.
- Analysis: Correlate the dramatic decrease in reaction rate with increasing n [9] to changes in the PES. Observe the shift from a double-well PES (gas phase) towards a unimodal PES (solution phase) as solvation stabilizes the charge-localized reactants more than the diffuse-charge transition state. This alters the dominance of indirect (complex-mediated) vs. direct reaction dynamics, indirectly influencing the feasibility of stereoelectronically constrained pathways.
Visualization of Concepts and Workflows
Diagram 1: Stereoelectronic Decision Tree for Reaction Pathways
Diagram 2: SN2 Trajectory Analysis and Pathway Separation
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Reagents and Computational Tools for Stereoelectronic Mechanism Studies
| Item Name / Category | Function / Purpose in Research | Example / Specification |
|---|---|---|
| Deuterated Substrates | Enable measurement of primary Kinetic Isotope Effects (KIEs) to probe C–H bond cleavage in the rate-determining step. | 1-Bromo-2,2-dideuterio-2-phenylethane [11]; synthetically prepared via reduction with LiAlD4. |
| Sterically-Hindered Strong Bases | To study the influence of base size on regiochemistry (E2) and competition between E2/SN2 pathways. | Potassium tert-butoxide (t-BuOK) [11]. |
| Crossed Molecular Beam Apparatus | For gas-phase reaction dynamics studies, providing control over collision energy and enabling measurement of product scattering angles. | Used in studies of F− + CH3Cl/I reactions to differentiate direct vs. indirect mechanisms [10]. |
| Ab Initio / DFT Software | To compute potential energy surfaces, locate transition states, calculate vibrational frequencies, and generate inputs for dynamics simulations. | Gaussian, ORCA, Q-Chem; used for benchmark studies of SN2/E2 barriers [9]. |
| Chemical Dynamics Simulation Code | To perform quasi-classical trajectory calculations on analytical or direct-dynamics PESs, revealing atomistic mechanisms and branching ratios. | VENUS, AMBER, CHARMM; employed to discover double-inversion pathways [10]. |
| Microsolvation Cluster Sources | To generate precisely solvated ionic nucleophiles (e.g., X−(H2O)_n) for studying the stepwise transition from gas-phase to solution-phase reactivity. | Supersonic expansion nozzles or ion guides; used in kinetics studies of Cl−(H2O)_n reactions [9]. |
| Isotope-Selective Detectors | To quantify the ratios of different isotopologues in reaction products or remaining substrates for accurate KIE determination. | Mass Spectrometry (MS) coupled with Gas Chromatography (GC-MS) or Liquid Chromatography (LC-MS). |
The backside displacement mechanism is a fundamental concept in organic chemistry, central to the stereospecificity of bimolecular nucleophilic substitution (SN2) reactions. This process, universally characterized by a complete inversion of configuration at the electrophilic carbon center, was first systematized through the discovery of the Walden inversion. For researchers investigating the contrasting frontside attack mechanisms (SN2X), a thorough understanding of the stereoelectronic constraints governing the traditional SN2 pathway is essential. This guide provides an in-depth technical examination of the inversion mechanism, its experimental validation, and critical quantitative data for professionals in reaction mechanism research and stereospecific drug development.
The SN2 Mechanism and Stereochemical Consequences
The Concerted Backside Attack
The SN2 reaction is a concerted, single-step process in which bond formation between the nucleophile and the electrophilic carbon occurs simultaneously with bond cleavage to the leaving group [3]. The reaction is termed "bimolecular" because its rate-determining step involves two molecular entities: the nucleophile and the substrate. The mechanism proceeds via a backside attack, where the nucleophile approaches the carbon center from the side directly opposite (180°) to the leaving group [3] [1]. This specific trajectory minimizes electronic repulsion between the incoming nucleophile and departing leaving group, and avoids steric hindrance from substituents on the carbon center.
During the transition state, the carbon atom adopts a trigonal bipyramidal geometry with partial bonds to both the nucleophile and leaving group [3]. The three non-participating substituents temporarily occupy a planar arrangement before the leaving group fully departs, causing these three groups to "flip" into an inverted spatial orientation—an process often analogized to an umbrella turning inside-out in strong wind [12] [3].
Inversion of Configuration
The stereochemical consequence of the backside attack is a complete inversion of configuration at the chiral center. If the starting substrate is a single enantiomer (e.g., the R-configuration), the product will be the opposite enantiomer (the S-configuration) [1]. This phenomenon is termed Walden inversion after Paul Walden, who first demonstrated this stereochemical transformation in 1896 [12] [13].
The SN2 reaction is stereospecific: different stereoisomers as substrates yield distinct stereoisomers as products. An R-enantiomer substrate produces exclusively the S-enantiomer product, while an S-enantiomer substrate yields exclusively the R-enantiomer product [1]. This specificity is a crucial consideration in pharmaceutical synthesis, where the biological activity of drug molecules often depends critically on their absolute configuration.
Table 1: Stereochemical Outcomes of Nucleophilic Substitution Mechanisms
| Reaction Parameter | SN2 Mechanism | SN1 Mechanism |
|---|---|---|
| Molecularity | Bimolecular | Unimolecular |
| Stereochemistry | Complete inversion | Racemization |
| Stereospecificity | Yes | No |
| Key Intermediate | Pentacoordinate transition state | Planar carbocation |
| Rate Equation | k[substrate][nucleophile] | k[substrate] |
Historical Context: The Walden Cycle
Paul Walden's seminal 1896 experiment established the phenomenon of inversion through a closed transformation sequence known as the Walden cycle [12] [13]. This series of reactions demonstrated the interconversion of enantiomers through processes involving both inversion and retention of configuration:
- (+)-chlorosuccinic acid (1) was converted to (+)-malic acid (2) using silver oxide (Ag₂O) in water, proceeding with retention of configuration [12].
- (+)-malic acid (2) was treated with phosphorus pentachloride (PCl₅) to yield the enantiomeric (-)-chlorosuccinic acid (3), proceeding with inversion of configuration [12] [13].
- (-)-chlorosuccinic acid (3) was converted to (-)-malic acid (4) using silver oxide, again with retention of configuration.
- Finally, (-)-malic acid (4) was treated with PCl₅ to return to the starting (+)-chlorosuccinic acid, completing the cycle with another inversion of configuration [12].
The net transformation of the cycle represented two inversions, resulting in overall retention, but critically demonstrated that certain reactions could flip the stereochemical configuration at a chiral center [12]. Modern computational studies reveal that the retention steps in the Walden cycle proceed through a double inversion mechanism involving formation of a β-lactone intermediate rather than direct substitution with retention [12].
Quantitative Analysis of SN2 Reactions
Structural and Solvent Effects on Reaction Rates
The rate of SN2 reactions is highly sensitive to structural features of the substrate, particularly the degree of substitution at the electrophilic carbon center. This structure-reactivity relationship arises from a combination of steric effects and electronic stabilization.
Table 2: Relative Rates of SN2 Reactions Based on Substrate Structure
| Substrate Type | Example Compound | Relative Rate | Structural Rationale |
|---|---|---|---|
| Methyl | CH₃Br | ~120,000,000 | Minimal steric hindrance to backside approach |
| Primary | CH₃CH₂Br | ~6,000,000 | Slight increase in steric hindrance |
| Secondary | (CH₃)₂CHBr | ~80,000 | Significant steric hindrance |
| Tertiary | (CH₃)₃CBr | ~0.0001 | Extreme steric hindrance prohibiting backside attack |
| Neopentyl | (CH₃)₃CCH₂Br | Extremely slow | β-branching creates severe steric shield |
The dramatic decrease in SN2 reactivity with increasing substitution reflects the steric congestion at the transition state [3]. In tertiary and neopentyl systems, the nucleophile cannot approach the backside of the carbon-leaving group bond without encountering severe van der Waals repulsions. Bridgehead systems such as 1-bromotriptycene are essentially inert to SN2 displacement due to complete blockade of the required backside approach trajectory [3].
Solvent and Leaving Group Effects
Polar aprotic solvents (e.g., DMSO, DMF, acetone) generally accelerate SN2 reactions by solvating cations effectively while leaving nucleophilic anions relatively "naked" and more reactive [3]. In contrast, protic solvents (e.g., water, alcohols) solvate nucleophiles through hydrogen bonding, reducing their nucleophilicity and slowing SN2 rates.
The nature of the leaving group also profoundly influences SN2 rates, with better leaving groups (those forming weaker bonds to carbon and stabilizing negative charge) accelerating reactions. The relative leaving group abilities generally follow the trend: Tosylate ≈ I⁻ > Br⁻ > Cl⁻ > F⁻.
Experimental Methodologies for Studying Inversion
Kinetic Analysis of SN2 Reactions
The definitive experimental evidence for the SN2 mechanism derives from kinetic studies showing second-order kinetics. The established protocol involves:
- Reaction Setup: Prepare separate solutions of the substrate (alkyl halide) and nucleophile in an appropriate solvent (often acetone or ethanol) [3].
- Rate Measurement: Monitor the disappearance of substrate or appearance of product over time using techniques such as conductivity measurement, GC, HPLC, or NMR spectroscopy.
- Order Determination: Determine the reaction order with respect to each reactant by maintaining one component in large excess while varying the concentration of the other.
- Kinetic Isotope Effects: Use isotopically labeled nucleophiles (e.g., radioiodide I⁻*) to trace the reaction pathway and quantify the rate of racemization versus substitution [3].
These kinetic analyses consistently demonstrate that the rate of the SN2 reaction is proportional to the concentrations of both the nucleophile and the substrate: Rate = k[substrate][nucleophile] [3].
Stereochemical Analysis Techniques
Determining the stereochemical course (inversion versus retention) requires starting with a substrate of known configuration and analyzing the product stereochemistry:
- Chiral Substrate Preparation: Obtain or synthesize an enantiomerically enriched substrate, typically a secondary alkyl halide or sulfonate with a known specific rotation [1].
- Nucleophilic Displacement: Conduct the substitution reaction under controlled conditions favoring SN2 mechanism (e.g., using strong nucleophile in polar aprotic solvent) [3].
- Product Analysis: Determine the configuration and enantiomeric purity of the product using polarimetry, chiral HPLC, or NMR with chiral shift reagents.
- Configuration Assignment: Compare the specific rotation of the product with known literature values to assign absolute configuration.
When (-)-2-bromooctane (R-configuration) reacts with hydroxide ion in an SN2 reaction, the product is (+)-2-octanol (S-configuration), demonstrating complete inversion of configuration [13].
Essential Research Reagents and Materials
Table 3: Key Research Reagents for Studying SN2 Mechanisms
| Reagent/Material | Function/Application | Technical Considerations |
|---|---|---|
| Alkyl Halides | Substrates of varying structure (methyl, primary, secondary, tertiary) | Purity essential; store under anhydrous conditions |
| Nucleophiles | Anionic species (I⁻, Br⁻, CN⁻, OH⁻) for displacement studies | Reactivity follows HSAB principles; may require phase-transfer catalysts |
| Polar Aprotic Solvents | DMSO, DMF, acetone for enhancing nucleophile reactivity | Must be rigorously dried; can penetrate skin (use appropriate PPE) |
| Silver Salts | Silver oxide (Ag₂O) used in Walden cycle experiments | Acts as hydroxide donor; light-sensitive |
| Phosphorus Pentachloride | Chlorinating agent for converting alcohols to alkyl chlorides | Moisture-sensitive; reacts violently with water |
| Chiral Auxiliaries | Resolving agents for obtaining enantiopure substrates | Essential for stereochemical studies |
| Deuterated Solvents | NMR analysis of reaction progress and stereochemistry | For mechanistic studies using NMR spectroscopy |
Visualizing the Backside Displacement Mechanism
Figure 1: Backside displacement mechanism showing inversion of configuration. The nucleophile approaches 180° from the leaving group, resulting in inversion of stereochemistry at the chiral center.
Figure 2: The Walden cycle demonstrating interconversion of enantiomers through alternating inversion and retention steps.
Implications for SN2X Reaction Mechanism Research
Understanding the stereoelectronic constraints governing the traditional SN2 mechanism provides critical context for ongoing research into frontside attack mechanisms (SN2X). While the classic SN2 pathway is stereospecific with complete inversion, the emerging class of halogenophilic (SN2X) and chalcogenophilic (SN2Ch) nucleophilic substitutions may proceed through different trajectories that enable frontside attack [14]. These alternative mechanisms potentially involve initial nucleophilic attack on the halogen or chalcogen atom rather than direct displacement at carbon.
For research scientists exploring these non-classical pathways, the established principles of Walden inversion serve as an essential benchmark. The experimental methodologies detailed herein—particularly kinetic isotope studies and stereochemical analysis—provide the foundational tools for distinguishing between backside displacement, frontside attack, and other mechanistic possibilities in nucleophilic substitution reactions.
The bimolecular nucleophilic substitution (SN2) reaction is a cornerstone of organic chemistry, characterized by a concerted mechanism in which bond formation and bond breaking occur simultaneously. The transition state (TS) of this reaction is a critical structure in which the central carbon atom becomes pentacoordinate, adopting a trigonal bipyramidal (TBP) geometry [15]. This geometry is not a stable intermediate but a fleeting, high-energy point on the reaction coordinate. This whitepaper provides an in-depth technical examination of the pentacoordinate carbon within the TBP geometry of the SN2 transition state. The analysis is framed within the context of ongoing research into the feasibility of "frontside attack" mechanisms, exploring the electronic and steric constraints that make the classical backside attack pathway dominant. Understanding the precise nature of bonding and geometry at this transition state is essential for researchers and drug development professionals manipulating reaction pathways in complex molecular systems, including pharmaceuticals.
The pursuit of a stable, or "frozen," SN2 transition state, representing a genuine pentacoordinate carbon species, remains an active area of computational and experimental research [16] [17] [18]. While common for silicon, stable pentacoordinate carbon compounds are exceptionally rare, prompting investigations into whether carbon can exhibit hypervalency. These studies probe the fundamental limits of carbon bonding and have significant implications for reaction design. Advanced analytical techniques, such as Atoms-in-Molecules (AIM) and Electron Localization Function (ELF) topology, are used to characterize the nature of the interactions in these unique systems [16].
Geometric and Electronic Structure of the Transition State
Trigonal Bipyramidal Geometry and Stereochemical Outcome
The SN2 transition state features a central carbon atom bonded to five other atoms in a trigonal bipyramidal (TBP) arrangement [3] [15]. In this geometry, the incoming nucleophile and the outgoing leaving group occupy the two axial positions, forming a linear Nu-C-LG axis. The three substituents originally attached to the electrophilic carbon reside in the equatorial plane, with bond angles of 120° [2]. This specific geometry is the direct cause of the stereospecific outcome of the SN2 reaction. The nucleophile must attack from the backside, 180° relative to the leaving group, for optimal orbital overlap [1] [15]. This backside attack forces the three equatorial groups to "flip" like an umbrella in a strong wind as the reaction proceeds through the transition state, resulting in an inversion of configuration (Walden inversion) at the chiral carbon center [3] [1].
Table 1: Key Geometric and Electronic Features of the SN2 Transition State
| Feature | Description | Implication for Mechanism |
|---|---|---|
| Coordination Number | Pentacoordinate Carbon | A fleeting, five-coordinate structure, not a stable intermediate [19]. |
| Molecular Geometry | Trigonal Bipyramidal (TBP) | Axial positions for nucleophile and leaving group; equatorial plane for the three original substituents [2] [15]. |
| Stereochemistry | Inversion of Configuration | A direct result of the backside attack and TBP geometry [3] [1]. |
| Reaction Kinetics | Second-Order (Bimolecular) | Rate = k[Nu:⁻][Substrate]; both species are involved in the rate-determining step [15]. |
| Bonding Nature | Non-Integer Electron Sharing (AIM/ELF) | Bond paths may exist, but electron count per "bond" is often significantly less than two [16]. |
Electronic Structure and Bonding Analysis
The electronic rearrangement during the SN2 reaction can be viewed as a HOMO-LUMO interaction, where the lone pair orbital of the nucleophile donates electrons into the σ* antibonding orbital of the carbon-leaving group (C–LG) bond [3] [15]. This interaction weakens the C–LG bond as the new C–Nu bond begins to form. At the transition state, the central carbon is approximately sp2-hybridized, with a p orbital forming during the transition to the product's molecular orbitals [15].
Advanced computational analyses provide deep insights into the nature of bonding at the pentacoordinate carbon. Atoms-in-Molecules (AIM) analysis often reveals bond critical points (BCPs) between the central carbon and all five surrounding atoms, suggesting the presence of an "interaction" [16]. However, the electron density, ρ(r), at these BCPs is typically about 0.02 atomic units, which is an order of magnitude lower than that of a standard C–C single bond (~0.28 au). This indicates these are weak interactions rather than full two-electron bonds [16].
Complementary Electron Localization Function (ELF) analysis can paint a different picture. For a proposed pentacoordinate carbon structure with a cyclopentadienyl anion and CN groups, ELF revealed only three disynaptic basins surrounding the central carbon, not five [16]. The total electron population in these basins was about 7.85 electrons, confirming that the central carbon is not hypervalent and possesses a familiar octet. This suggests that while the carbon is pentacoordinate from a geometric perspective, it is not pentavalent in the classical Lewis sense, with the axial interactions being notably weaker and involving fewer electrons [16].
Diagram 1: SN2 reaction pathway and transition state geometry.
Computational Protocols for Characterizing Transition States
Locating and Verifying the Transition State
Computational chemistry is indispensable for studying SN2 transition states due to their transient nature. Several robust protocols exist for locating and characterizing these saddle points on the potential energy surface.
Eigenvector Following (Manual Guess): This method requires an initial guess of the transition state structure, which is then optimized using first and second derivatives. For an SN2 reaction, a chemist can build a structure with a TBP geometry where the C–Nu and C–LG distances are elongated and equal. The optimization follows the eigenvector with a negative eigenvalue (the reaction coordinate). It is efficient to start this process by calculating the Hessian (matrix of second derivatives) once at the beginning (
Opt=CalcFCin Gaussian) [20]. This approach works well for simple SN2 systems where a reasonable TS guess can be constructed.Synchronous Transit Methods (QST2/QST3): These methods are highly effective for bimolecular reactions like SN2. They require the structures of the reactant complex and the product complex.
- QST2: The input file contains the optimized structures of the reactant and product complexes. The algorithm performs a linear or quadratic interpolation between them to generate an initial guess for the TS, which is then optimized [20].
- QST3: This more robust method requires the reactant complex, product complex, and a user-supplied guess for the TS structure. It overcomes limitations of poor interpolated guesses in QST2 [20]. A typical workflow for a Menshutkin reaction (a type of SN2 reaction) at the HF/3-21G level using QST2 might yield a TS with C–N and C–Cl distances of ~1.95 Å and ~2.42 Å, respectively [20].
Potential Energy Surface (PES) Scanning: A relaxed PES scan is performed by constraining a key internal coordinate (e.g., the C–LG distance) and optimizing all other degrees of freedom at each point. The maximum energy point along the scan provides a structure close to the true TS, which can then be fully optimized and verified. This method is reliable but computationally demanding [20].
Verification and Energy Calculation
After a stationary point is located, a frequency calculation is mandatory to confirm it is a first-order saddle point (a transition state) and not a minimum. The key indicator is the presence of one, and only one, imaginary vibrational frequency (negative eigenvalue). The normal mode of this imaginary frequency should correspond to the expected reaction coordinate—the simultaneous formation of the C–Nu bond and breaking of the C–LG bond [20].
Once the TS is verified, single-point energy calculations at a higher level of theory (e.g., CCSD(T)/cc-pVTZ) on the pre-optimized MP2/cc-pVTZ structures can provide accurate activation energies (Ea) and reaction energies (ΔE) [20].
Table 2: Summary of Key Computational Protocols for SN2 Transition State Optimization
| Method | Key Input Requirements | Advantages | Limitations |
|---|---|---|---|
| Eigenvector Following | A single, reasonable guess for the TS structure. | Fast for simple systems; intuitive. | Requires a good initial guess; can fail for complex reactions. |
| QST2 | Optimized structures of the reactant and product complexes. | Automated; no need for a TS guess. | Can fail if the interpolation produces a chemically unreasonable structure. |
| QST3 | Optimized reactant, product, and a guess TS structure. | More robust and reliable than QST2. | Requires building three structures. |
| PES Scanning | A defined internal coordinate to scan (e.g., C-LG distance). | Highly reliable; maps the reaction path. | Computationally expensive for large systems or multiple variables. |
The Challenge of "Frozen" Transition States and Frontside Attacks
Pursuing a Stable Pentacoordinate Carbon
A significant research endeavor is the computational and experimental design of molecules that mimic the SN2 transition state as a stable minimum on the potential energy surface, not a saddle point. Success in this area would demonstrate viable pentacoordinate carbon. Key strategies and findings include:
Electronegative Substituents and Large Leaving Groups: Bickelhaupt and co-workers proposed a series of compounds, X–C(CN)₃–X⁻ (X = Br, I, At), where the highly electronegative CN groups help stabilize a planar CR₃ radical motif, and the large halogens (especially At) allow for a stable D3h-symmetric structure that resembles a "frozen" SN2 TS [17]. The stability of this structure is highly sensitive to the computational method and the identity of the halogen.
Carbon-Carbon Bonding Environments: Rzepa proposed a system using the aromatic cyclopentadienyl anion as a large nucleophile/leaving group, creating a pentacoordinate carbon with five C–C interactions [18]. A vibrational frequency calculation on this system found a real, positive frequency for the typical "umbrella" SN2 mode, indicating a stable minimum, not a transition state [18].
The C(CH₃)₅⁺ Cation: Schleyer and Schaefer investigated the pentamethylmethane cation, C(CH₃)₅⁺, which was a local minimum with five bond critical points from the central carbon. However, the axial bonds were very long (~1.736 Å), and the dissociation barriers were extremely low (~1.5 kcal/mol), indicating spontaneous dissociation and making isolation impractical [21].
Implications for Frontside Attack Mechanisms
The classical SN2 reaction proceeds exclusively via backside attack. The search for stable pentacoordinate carbon and the analysis of its bonding have direct implications for the feasibility of frontside attack mechanisms.
The AIM and ELF analyses of stabilized SN2-like structures reveal that the axial "bonds" are weak interactions with low electron density, not full covalent bonds [16]. For a frontside attack to be competitive, the nucleophile would have to approach the same side as the leaving group, which is both sterically blocked and electronically repulsive, as both the nucleophile and leaving group are electron-rich. The stable "frozen" TS structures achieve their stability through geometric constraints, ionic interactions, and highly delocalized electron systems that mitigate these repulsions, but they do not represent a simple two-electron bond formation from the front side. The evidence suggests that a concerted frontside displacement leading to retention of configuration remains highly unfavorable compared to the low-energy pathway offered by backside attack and TBP inversion.
Diagram 2: Computational workflow for transition state characterization.
The Scientist's Toolkit: Essential Reagents and Computational Methods
Table 3: Research Reagent Solutions for SN2 Transition State Analysis
| Reagent / Method | Type | Function in Research |
|---|---|---|
| Astatine (At) / Large Halogens | Chemical Element | Used as a leaving group (X) in proposed "frozen" TS molecules (e.g., X–C(CN)₃–X⁻). Its large size and polarizability help stabilize the pentacoordinate carbon structure [17] [18]. |
| Cyclopentadienyl Anion | Organic Anion | Acts as a bulky, aromatic nucleophile/leaving group (X) in model systems to create a pentacoordinate carbon center with five C–C interactions for study [16] [18]. |
| Nitrile (CN) Groups | Functional Group | Electronegative substituents (Y) that help stabilize a planar CR₃ radical center and withdraw electron density, aiding in the stabilization of the central carbon in hypercoordinate systems [17]. |
| Atoms-in-Molecules (AIM) | Computational Analysis | Topological analysis of the electron density to locate bond critical points (BCPs), providing a geometric criterion for the existence of a bond/interaction [16]. |
| Electron Localization Function (ELF) | Computational Analysis | Partitions space into basins to analyze electron pairing and localization. Used to determine the number of electrons associated with a bond, distinguishing hypercoordination from hypervalency [16]. |
Experimental Evidence for Stereospecificity in SN2 Reactions
The bimolecular nucleophilic substitution (SN2) reaction represents a fundamental transformation in organic chemistry, characterized by a concerted mechanism that results in the inversion of stereochemical configuration at the carbon reaction center. This in-depth technical guide examines the foundational experimental evidence establishing the stereospecific nature of the classic backside attack SN2 mechanism, while framing these established principles within the emerging context of frontside attack nucleophilic substitution (SN2X) reaction mechanism research. By synthesizing classical kinetic studies, stereochemical investigations, and contemporary research breakthroughs, this review provides researchers, scientists, and drug development professionals with a comprehensive experimental framework for understanding and applying stereospecific substitution principles in complex molecular settings.
The SN2 (substitution nucleophilic bimolecular) mechanism represents one of the most thoroughly studied and fundamental reaction pathways in organic chemistry. First characterized in the 1930s by Hughes and Ingold, this concerted process involves the simultaneous bond formation between a nucleophile and an electrophilic carbon center with bond cleavage between that carbon and a leaving group [3] [22]. The mechanism is characterized by its bimolecular nature, with reaction rates dependent on both nucleophile and substrate concentrations, following second-order kinetics [23] [24]. The notation "SN2" specifically denotes Substitution, Nucleophilic, and bimolecular, reflecting the participation of two molecular entities in the rate-determining step [3].
A defining characteristic of the traditional SN2 mechanism is its stereochemical outcome. The reaction proceeds via a concerted backside attack, wherein the nucleophile approaches the carbon center 180° opposite the departing leaving group, resulting in inversion of configuration at the stereocenter [3]. This stereospecific process, often termed Walden inversion, has been considered a cornerstone of mechanistic organic chemistry for nearly a century [3]. The transition state for this reaction features a trigonal bipyramidal geometry in which the nucleophile and leaving group form partial bonds with the central carbon, while the three remaining substituents adopt a coplanar arrangement [3].
Recent research has revealed unexpected complexity in this seemingly settled mechanistic paradigm with the discovery of a novel frontside attack mechanism (designated SN2X) that challenges the exclusive dominance of the backside attack pathway under specific circumstances [22]. This review examines the definitive experimental evidence supporting the stereospecific nature of the classical SN2 mechanism while contextualizing these findings within the broader landscape of nucleophilic substitution research, including contemporary investigations into non-traditional reaction pathways.
Experimental Evidence for Stereospecificity
Kinetic Evidence for a Concerted Bimolecular Mechanism
The foundational evidence for the SN2 mechanism derives from kinetic studies that established its distinct second-order rate law. Unlike stepwise mechanisms that may depend only on substrate concentration, the SN2 reaction rate exhibits a direct dependence on both nucleophile and substrate concentrations [23] [24]. This relationship is expressed mathematically in the rate equation:
Rate = k[Substrate][Nucleophile]
where k represents the rate coefficient or rate constant [23] [24]. This second-order kinetic profile provides the initial evidence for a mechanism involving both reaction components in the rate-determining step, consistent with a concerted process without stable intermediates.
The experimental determination of this rate law typically involves systematic variation of reactant concentrations while monitoring reaction progress through techniques such as conductivity measurements, spectrophotometry, or chromatography. For example, in the reaction of hydroxide ion with bromomethane, doubling either reactant concentration produces a corresponding doubling of the reaction rate, confirming the bimolecular nature of the process [25].
Stereochemical Evidence: Inversion of Configuration
The most definitive evidence for the backside attack mechanism comes from stereochemical studies using chiral substrates. When a nucleophile reacts with a stereodefined chiral substrate bearing a leaving group at the stereocenter, the SN2 mechanism produces a product with inverted configuration [3].
A classic experimental demonstration involves the reaction of (S)-2-bromobutane with hydroxide ion to form 2-butanol. The reaction proceeds with clean inversion of configuration, yielding exclusively (R)-2-butanol rather than a racemic mixture [3]. This stereochemical outcome provides compelling evidence for a concerted mechanism with nucleophilic attack from the side opposite the departing leaving group.
The stereospecificity of the SN2 reaction is further demonstrated through Walden cycle experiments, wherein a series of transformations beginning and ending with the same compound nonetheless result in net inversion of configuration due to an odd number of SN2 steps within the sequence [3]. This phenomenon, first observed by Paul Walden in 1896, provides additional corroborating evidence for the inversion pathway [3].
Figure 1: Stereochemical inversion in the SN2 reaction of (S)-2-bromobutane to form (R)-2-butanol
Steric and Substrate Effects on Reactivity
The sensitivity of SN2 reactions to steric hindrance provides additional mechanistic evidence. Reaction rates decrease dramatically as substitution at the carbon reaction center increases, consistent with the steric requirements of backside attack [25]. Experimental relative reactivity data demonstrate this trend:
Table 1: Relative SN2 Reaction Rates by Substrate Class
| Substrate Class | Example | Relative Rate |
|---|---|---|
| Methyl | CH₃Br | ~1200 [25] |
| Primary | CH₃CH₂Br | ~40 [25] |
| Secondary | (CH₃)₂CHBr | ~1 [25] |
| Tertiary | (CH₃)₃CBr | Too slow to measure [25] |
| Neopentyl | (CH₃)₃CCH₂Br | Extremely slow [25] |
This reactivity pattern reflects the increasing difficulty of nucleophile approach as substituents at the reaction center become more numerous and bulky. The virtual absence of SN2 reactivity for tertiary and neopentyl substrates provides indirect but compelling evidence for the sterically constrained backside attack pathway [25].
Nucleophile Reactivity Trends
The correlation between nucleophile strength and SN2 reaction rates further supports the concerted mechanism. Unlike stepwise processes where nucleophile strength may have minimal impact on rate, SN2 reactions show pronounced sensitivity to nucleophile identity and concentration [25]. Experimental data demonstrate significant rate variations across different nucleophiles:
Table 2: Relative Rates of SN2 Reactions with Bromomethane by Nucleophile
| Nucleophile | Product | Relative Rate |
|---|---|---|
| H₂O | CH₃OH₂⁺ | 1 [25] |
| CH₃CO₂⁻ | CH₃CO₂CH₃ | 500 [25] |
| NH₃ | CH₃NH₃⁺ | 700 [25] |
| Cl⁻ | CH₃Cl | 1,000 [25] |
| HO⁻ | CH₃OH | 10,000 [25] |
These reactivity trends reflect the importance of both nucleophile strength and steric accessibility in the SN2 mechanism, consistent with direct participation of the nucleophile in the rate-determining step through backside attack.
Experimental Protocols for Demonstrating Stereospecificity
Kinetic Rate Determination Protocol
Objective: Determine the kinetic order of a nucleophilic substitution reaction with respect to both substrate and nucleophile.
Materials:
- Alkyl halide substrate (e.g., bromomethane, 2-bromobutane)
- Nucleophile (e.g., sodium hydroxide, potassium iodide)
- Appropriate solvent (e.g., aqueous ethanol, acetone)
- Conductivity meter or UV-Vis spectrophotometer
- Thermostatted reaction vessel
Methodology:
- Prepare a series of reaction mixtures with constant nucleophile concentration while varying substrate concentration (0.01 M to 0.1 M range)
- Monitor reaction progress by measuring conductivity changes or absorbance at appropriate wavelengths
- Determine initial rates from the linear portion of concentration versus time plots
- Repeat the experiment with constant substrate concentration while varying nucleophile concentration
- Construct logarithmic plots of initial rate versus concentration to determine reaction orders
Expected Results: A linear plot of log(rate) versus log[substrate] with slope of 1.0, and log(rate) versus log[nucleophile] with slope of 1.0, confirming second-order kinetics consistent with the SN2 mechanism [23] [24].
Stereochemical Inversion Demonstration Protocol
Objective: Demonstrate inversion of configuration in the SN2 reaction using a chiral substrate.
Materials:
- Optically active (S)-2-bromobutane
- Sodium hydroxide solution
- Absolute ethanol
- Polarimeter
- Separation funnel and rotary evaporator
Methodology:
- Dissolve (S)-2-bromobutane in ethanol and record initial optical rotation
- Add standardized sodium hydroxide solution with stirring
- Monitor reaction completion by TLC or GC
- Extract and purify the resulting 2-butanol product
- Measure the specific rotation of the product using a polarimeter
- Compare the magnitude and sign of optical rotation with authentic samples of (R)- and (S)-2-butanol
Expected Results: The product exhibits optical rotation opposite in sign but approximately equal in magnitude to the starting material, confirming stereospecific inversion of configuration [3].
Steric Hindrance Investigation Protocol
Objective: Quantitatively demonstrate the effect of substrate sterics on SN2 reactivity.
Materials:
- Series of alkyl halides (methyl, primary, secondary, tertiary)
- Standardized sodium iodide in acetone
- UV-Vis spectrophotometer or GC-MS
- Constant temperature bath
Methodology:
- Prepare solutions of each alkyl halide in acetone at identical concentrations
- Add equal volumes of sodium iodide in acetone to initiate reactions
- Monitor reaction progress by measuring appearance of halide ion or disappearance of substrate
- Determine pseudo-first-order rate constants for each substrate under identical conditions
- Compare relative reaction rates across the substrate series
Expected Results: A significant decrease in reaction rate with increasing substitution at the reaction center, demonstrating the steric requirements of the SN2 mechanism [25].
The SN2X Reaction: Frontside Attack Mechanism
Discovery and Mechanism
Recent research has challenged the long-standing dogma that SN2 reactions exclusively proceed via backside attack. In 2019, Professor Choon-Hong Tan and colleagues at Nanyang Technological University reported a novel halogenophilic nucleophilic substitution (SN2X) reaction that proceeds through a frontside attack mechanism [22]. This unprecedented pathway involves nucleophile approach from the same side as the leaving group, rather than the opposite side characteristic of traditional SN2 reactions.
The SN2X mechanism appears to be facilitated by specific substrate features, particularly the presence of halogens at the reaction center that can interact favorably with the incoming nucleophile during the frontside approach [22]. This discovery emerged from careful experimental design that systematically eliminated alternative reaction pathways and confirmed the unique stereochemical outcome inconsistent with backside attack.
Figure 2: Comparison of traditional SN2 backside attack and the novel SN2X frontside attack mechanisms
Implications for Stereospecificity
The discovery of the SN2X mechanism necessitates a refined understanding of stereospecificity in nucleophilic substitution. While the traditional SN2 reaction remains stereospecific with inversion of configuration, the SN2X pathway may exhibit different stereochemical outcomes or altered stereospecificity patterns [22]. This breakthrough suggests that the stereochemical course of nucleophilic substitution reactions may be more complex than previously recognized, with potential alternative pathways available under specific circumstances.
Contemporary research continues to explore the boundaries and applications of this novel mechanism. Recent investigations have demonstrated stereospecific nucleophilic substitution at quaternary carbon stereocenters using cyclopropyl carbinol derivatives, expanding the potential synthetic utility of stereospecific substitution reactions at highly congested centers [26] [27]. These developments highlight the ongoing evolution of our understanding of nucleophilic substitution mechanisms and their stereochemical consequences.
Advanced Applications and Research Tools
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Stereospecific Substitution
| Reagent | Function | Application Example |
|---|---|---|
| Optically active alkyl halides | Chiral substrates | Stereochemical fate studies [3] |
| Sodium iodide in acetone | Nucleophile source | Halide reactivity comparisons [25] |
| D₂O or H₂¹⁸O | Isotopically labeled nucleophiles | Mechanistic tracer studies |
| Polarimeter | Optical rotation measurement | Stereochemical outcome determination [3] |
| Chiral shift reagents | NMR enantiomeric differentiation | Product configuration analysis |
Computational Approaches
Modern computational methods provide additional tools for investigating SN2 stereospecificity. Density functional theory (DFT) and coupled-cluster calculations allow researchers to model reaction transition states, quantify activation barriers, and predict stereochemical outcomes [28]. These approaches have been particularly valuable in studying the novel SN2X mechanism and exploring its relationship to traditional SN2 pathways.
Computational analyses have revealed that the competition between reaction pathways can be understood through concepts such as "characteristic distortivity" and "transition state acidity," providing theoretical frameworks for predicting and controlling substitution outcomes [28]. These tools have become indispensable for mechanistic studies in contemporary research settings.
The experimental evidence for stereospecificity in SN2 reactions represents a cornerstone of mechanistic organic chemistry. Kinetic studies, stereochemical investigations, and steric effect analyses collectively provide compelling support for the classic backside attack mechanism with inversion of configuration. These established principles continue to inform synthetic design and mechanistic analysis in both academic and industrial settings.
The recent discovery of the frontside attack SN2X mechanism demonstrates that fundamental reaction pathways can still yield surprising revelations. This finding not only expands the synthetic toolbox available to chemists but also illustrates the importance of continued rigorous investigation of even seemingly settled mechanistic questions. The integration of traditional experimental approaches with modern computational methods provides a powerful framework for further elucidating the complexities of nucleophilic substitution reactions.
For researchers in pharmaceutical development and complex molecule synthesis, understanding both the classical principles of SN2 stereospecificity and the emerging possibilities of alternative mechanisms offers opportunities for innovative synthetic strategies. As research in this area continues to evolve, further surprises and applications will undoubtedly emerge, reinforcing the dynamic nature of organic chemistry as a discipline.
Potential Energy Surface Profiles for Concerted Substitution Mechanisms
The Potential Energy Surface (PES) provides a fundamental theoretical framework for understanding the energetic pathways and kinetic outcomes of chemical reactions. In the context of nucleophilic substitution reactions, the PES represents the potential energy of the system as a function of the relative positions of all atoms involved during the reaction pathway. For concerted substitution mechanisms—particularly the SN2 reaction and its variants—the PES reveals a single energy barrier corresponding to the transition state where bond formation and bond breaking occur simultaneously. Recent investigations have revealed that the mechanism of aliphatic nucleophilic reactions may not be as straightforward as once thought, with some SN2 processes potentially proceeding via front-side attack or involving intermediate complexes [29]. This technical guide examines the PES characteristics of concerted substitution mechanisms within the broader research context of frontside attack SN2X reaction mechanisms, providing researchers with both foundational principles and advanced methodological approaches for investigating these fundamental chemical transformations.
Fundamental PES Characteristics for SN2 Reactions
Classical SN2 Reaction Coordinate Diagram
The classical SN2 reaction proceeds through a concerted mechanism in which nucleophile attack and leaving group departure occur simultaneously through a single transition state [30] [3]. The PES for this reaction displays characteristic features that distinguish it from stepwise mechanisms:
- Single Energy Barrier: The reaction pathway encounters only one maximum corresponding to the transition state structure [30]
- Transition State Geometry: At the energy maximum, the central carbon exhibits a trigonal bipyramidal geometry with partial bonds to both nucleophile and leaving group [3]
- Stereochemical Outcome: The backside attack trajectory results in inversion of configuration at the electrophilic carbon center [3]
- Kinetic Order: The reaction follows second-order kinetics, with rate dependent on both nucleophile and substrate concentrations [3]
The energy diagram for the SN2 reaction between CH₃Br and OH⁻ demonstrates that products CH₃OH and Br⁻ exist at lower energy than reactants, indicating an exothermic process with a single transition state at the energy maximum [30].
Structural and Electronic Factors Influencing the PES
The energy landscape of SN2 reactions is significantly influenced by the structure of the alkyl halide substrate and electronic effects:
Table 1: SN2 Reaction Rates Based on Alkyl Halide Structure
| Alkyl Halide Type | Structure | Relative Rate |
|---|---|---|
| Methyl | CH₃-X | 30 |
| Primary | R-CH₂-X | 1 |
| Secondary | R₂CH-X | 0.03 |
| Tertiary | R₃C-X | Too slow to measure |
Data compiled from experimental kinetic studies [30]
This dramatic rate reduction with increasing substitution results from steric hindrance that impedes the backside approach of the nucleophile to the electrophilic carbon center [30]. Additionally, electronic effects from substituents on the tetrahedral carbon can substantially alter activation energies, which range from 0.3 to 23.5 kcal·mol⁻¹ across different substrates [31].
Advanced PES Features in Non-Classical SN2 Mechanisms
Contact Ion-Pair SN2 Mechanisms
Recent experimental investigations using ¹⁹F-NMR spectroscopy have revealed a contact ion-pair mechanism that contrasts with conventional SN2 perspectives [29]. In this novel mechanism:
- The nucleophile and counter-cation remain in close contact throughout the reaction
- Lewis base phase transfer catalysts coordinate with the cation, effectively mitigating its retarding Coulomb forces on the nucleophile
- The nucleophile behaves as an "almost bare" entity despite the CIP configuration
- This mechanism accounts for the remarkable efficiency of SN2 processes using alkali metal salts with appropriate catalysts
This CIP mechanism represents a significant departure from the traditional "solvent-separated ion pair" conception and provides new dimensions to the PES of SN2 reactions [29].
Electron Density Perspectives from MEDT
Within the Molecular Electron Density Theory framework, analysis of the electron localization function provides profound insights into the electronic reorganization along the reaction coordinate:
- Transition states in symmetric SN2 reactions are characterized by ionic species rather than hypervalent carbon
- The electronic similarity between SN1 and SN2 molecular mechanisms suggests leaving group departure may occur before nucleophile approach in certain cases
- Relative Interacting Atomic Energy analysis enables decomposition of electronic factors responsible for activation energies
- The electrophilicity index of substrates correlates with their tendency to participate in polar reactions with nucleophiles [31]
These MEDT approaches provide a more nuanced understanding of the electronic factors shaping the PES of substitution reactions beyond conventional orbital-based rationalizations.
Methodological Approaches for PES Investigation
Computational Protocol for PES Mapping
Table 2: Methodologies for PES Determination in Substitution Reactions
| Method | Application | Key Outputs | Considerations |
|---|---|---|---|
| MEDT with ELF/RIAE | Electron density evolution along reaction path | Bonding evolution theory, atomic energy contributions | Provides chemical intuition for electron reorganization |
| Differentiable Molecular Dynamics | PES refinement using experimental data | Transport coefficients, spectroscopic predictions | Combines computational efficiency with experimental accuracy |
| Machine Learning Potentials | High-dimensional PES fitting | IR and Raman spectra, reaction dynamics | Accuracy limited by underlying ab initio methods |
| Generalized Internal Coordinates | Vibrational structure determination | Optimized coordinate systems, vibrational transitions | Superior to normal hyperspherical coordinates for certain systems |
Methodologies compiled from recent literature [32] [31] [33]
Experimental Probes for Reaction Mechanism Elucidation
Advanced spectroscopic techniques provide crucial experimental validation for computational PES predictions:
- ¹⁹F-NMR Spectroscopy: Fingerprinting tool for identifying contact ion-pair complexes in SN2 reactions; chemical shifts at -123 to -121 ppm indicate CIP forms of KF and CsF [29]
- Kinetic Isotope Effects: Probe for transition state structure and bonding changes
- Stereochemical Analysis: Inversion of configuration confirms backside attack pathway [3]
- Cross-Over Experiments: Distinguish between intra- and intermolecular pathways
These experimental approaches provide essential data for refining computational PES models toward higher accuracy.
PES Characteristics of Frontside Attack SN2X Mechanisms
The emerging research on frontside attack mechanisms reveals distinctive PES features compared to classical SN2 pathways:
- Higher Energy Transition State: Frontside approach experiences greater steric and electronic repulsion
- Altered Stereochemical Outcome: Potential for retention of configuration rather than inversion
- Cation Assistance: Lewis acid catalysts may stabilize frontside transition states through coordination
- Solvent Dependence: Polar aprotic solvents may favor alternative reaction coordinates
Recent theoretical studies suggest that both electronic effects of substituents and the nature of the leaving group can shift the molecular mechanism of SN reactions from SN2 to SN1 pathways, with potential for intermediate mechanistic regimes [31].
Visualization of PES Profiles
Classical SN2 Reaction Coordinate
Diagram 1: SN2 Reaction Coordinate (6.1)
Contact Ion-Pair SN2 Mechanism
Diagram 2: Contact Ion-Pair Mechanism (6.2)
Research Reagent Solutions
Table 3: Essential Reagents for SN2 Mechanism Studies
| Reagent/Catalyst | Function | Application Context |
|---|---|---|
| 18-Crown-6 Ether | Lewis base phase transfer catalyst | Coordinates K⁺ to enhance fluoride nucleophilicity |
| [2,2,2]-Cryptand | Cation chelating agent | Creates "naked" nucleophile by complete cation encapsulation |
| Pentaethylene Glycol | Hydrogen-bonding Lewis base PTC | Activates CIP via OH coordination to cation |
| BINOL-based PentaEG | Chiral Lewis base PTC | Enables asymmetric induction in SN2 reactions |
| Alkali Metal Fluorides | Nucleophile source | MF (M = K, Cs) in CIP mechanisms |
| Tetraalkylammonium Salts | Traditional PTC catalysts | Provides bulky, diffuse counter-cations for nucleophiles |
Reagent functions compiled from experimental studies [29]
The investigation of Potential Energy Surface profiles for concerted substitution mechanisms continues to evolve beyond the classical SN2 paradigm toward more sophisticated understandings that incorporate ion-pairing, frontside attack pathways, and electron density-based analyses. The integration of advanced computational methods with experimental spectroscopic probes enables researchers to refine PES models with increasing accuracy, revealing continuum mechanisms rather than discrete pathways. For drug development professionals, these insights provide fundamental principles for predicting reactivity patterns and stereochemical outcomes in synthetic transformations, ultimately facilitating more efficient design of small molecule therapeutics. Future research directions will likely focus on further elucidating the role of solvent dynamics, cation effects, and non-covalent interactions in shaping the energy landscapes of these essential organic transformations.
Analytical Methods and Computational Approaches for Studying Substitution Mechanisms
Kinetic Analysis Techniques for Distinguishing Reaction Mechanisms
Kinetic analysis serves as a fundamental tool in physical organic chemistry for determining reaction rate laws and elucidating detailed reaction mechanisms. For researchers investigating novel pathways such as the frontside attack nucleophilic substitution (SN2X) mechanism, a suite of advanced techniques enables the discrimination between competing mechanistic models. This whitepaper provides an in-depth examination of Reaction Progress Kinetic Analysis (RPKA) and associated methodologies that allow scientists to probe reactions under synthetically relevant conditions, moving beyond traditional pseudo-first-order approaches to capture more representative catalytic behavior and identify subtle mechanistic distinctions critical to pharmaceutical development.
Chemical kinetics involves the study of reaction rates and the factors influencing them, providing the experimental foundation for proposing reaction mechanisms [34]. A reaction mechanism represents a theoretical model that explains not just the stoichiometric pathway but the precise series of elementary steps through which reactants transform into products [34]. For researchers exploring non-classical nucleophilic substitution pathways like the SN2X mechanism, kinetic analysis offers the primary experimental evidence to distinguish between conventional SN2 backside attack, SN1 unimolecular dissociation, and frontside attack pathways. Unlike stoichiometric mechanisms that merely show the sequence of steps, intimate mechanisms detail the relative positions of all atoms throughout the reaction, providing the level of detail required to confirm a novel mechanism [34].
Traditional approaches often rely on pseudo-first-order analysis using large excesses of reagents to simplify rate laws. However, the field has increasingly adopted Reaction Progress Kinetic Analysis (RPKA), formalized by Professor Donna Blackmond, which probes reactions at synthetically relevant concentrations and ratios [35]. This methodology provides more representative data about reaction behavior under actual synthetic conditions and can reveal subtle mechanistic features such as induction periods, catalyst deactivation, or changes in mechanism that might be obscured in traditional kinetics [35]. For pharmaceutical researchers investigating novel substitution mechanisms, these insights are crucial for understanding and optimizing catalytic reactions that underpin modern drug synthesis.
Core Kinetic Analysis Techniques
Reaction Progress Kinetic Analysis (RPKA)
RPKA represents a significant advancement over traditional kinetic methods by examining reactions where multiple reactant concentrations change measurably throughout the reaction course, rather than using overwhelming excesses of reagents [35]. This approach provides kinetic data under conditions more relevant to actual synthetic applications, where reagent concentrations are typically balanced. The methodology relies on accurately monitoring reaction conversion over time through various in situ techniques, then manipulating and presenting the data to reveal underlying rate laws and mechanistic features [35]. A key advantage of RPKA is its ability to identify unexpected kinetic behavior such as autocatalysis, catalyst degradation, or changes in rate-determining steps that might be missed under traditional pseudo-first-order conditions.
Essential Monitoring Techniques for Kinetic Studies
Various instrumental techniques enable the precise monitoring of reaction progress necessary for rigorous kinetic analysis:
Table 1: Key Techniques for Monitoring Reaction Kinetics
| Technique | Principle of Operation | Data Type | Applications in Mechanism Elucidation | Advantages | Limitations |
|---|---|---|---|---|---|
| Reaction Progress NMR | Tracks changes in integration of distinctive reactant/product peaks relative to internal standard [35] | Integral (concentration vs. time) [35] | Identification of intermediates in Buchwald-Hartwig amination [35] | Can identify species in solution; variable temperature capability [35] | Requires homogeneous systems; distinct NMR signals needed [35] |
| In situ FT-IR | Monitors changes in IR absorbance of functional groups via Beer's Law [35] | Integral (concentration vs. time) [35] | Mechanism of amido-thiourea catalyzed asymmetric Strecker synthesis [35] | Excellent for functional group tracking; modern deconvolution capabilities [35] | Spectral overlap can complicate analysis [35] |
| In situ UV-vis | Measures absorbance changes in UV/visible region via Beer's Law [35] | Integral (concentration vs. time) [35] | Study of samarium Barbier reaction [35] | Sensitive for chromophores; good for organometallic complexes [35] | Limited to systems with UV-active species [35] |
| Reaction Calorimetry | Monitors instantaneous heat flux proportional to reaction enthalpy change [35] | Differential (rate vs. time) [35] | Catalyst screening; prolinate-catalyzed α-amination [35] | Directly measures rate; no chromophores required [35] | Requires known reaction enthalpy [35] |
Data Manipulation and Presentation in Kinetic Analysis
The raw data obtained from monitoring techniques must be appropriately processed to extract meaningful kinetic information:
- Concentration vs. Time Plots: The most fundamental representation, showing [A]t versus time (t) for reactants and products [35].
- Fractional Conversion Plots: Data normalized using F = ([A]₀ - [A]t)/[A]₀, enabling comparison of reactions run at different initial concentrations [35].
- Reaction Rate vs. Time Plots: Particularly for calorimetric data, where rate v = q/(V·ΔH) with q representing instantaneous heat transfer, ΔH as reaction enthalpy, and V as reaction volume [35].
- Rate vs. Concentration Plots: Combined plots showing reaction rate (v) versus substrate concentration ([S]), where reaction progress reads from right to left along the x-axis [35].
For catalytic reactions specifically, kinetic analysis helps distinguish between steady-state conditions (where catalyst-substrate complex concentration remains low and constant) and pre-equilibrium conditions (with rapid, reversible substrate binding before a slow product-forming step) [35]. These distinctions directly impact the mathematical form of the rate law and provide evidence for the catalyst resting state, which is crucial for mechanistic assignment.
Experimental Protocols for Kinetic Analysis
General Workflow for Reaction Progress Kinetic Analysis
The following diagram outlines the comprehensive workflow for conducting RPKA studies:
Detailed Protocol: In situ FT-IR Kinetic Monitoring
Purpose: To determine the rate law of a nucleophilic substitution reaction by tracking the disappearance of a specific functional group using in situ Fourier Transform Infrared spectroscopy.
Materials and Equipment:
- FT-IR spectrometer with reaction chamber and temperature control
- Appropriate IR-transparent windows (e.g., KBr, NaCl)
- Reaction vessel compatible with in situ monitoring
- Syringes for reagent introduction
- Data acquisition software
Procedure:
- Method Development:
- Identify distinctive IR absorbance peaks for key reactants, products, or potential intermediates
- Establish calibration curves correlating absorbance with concentration for quantitative analysis
- Determine optimal data collection frequency to capture kinetic profile (typically 1-30 second intervals)
Reaction Initiation:
- Load reaction vessel with appropriate solvents and nucleophile
- Equilibrate to desired reaction temperature with continuous stirring
- Establish background spectrum under reaction conditions
- Rapidly introduce electrophile to initiate reaction while commencing data collection
Data Collection:
- Collect spectra at predetermined intervals throughout reaction
- Monitor isosbestic points to verify clean conversion or detect intermediates
- Continue data collection until reaction reaches >95% completion or rate becomes immeasurably slow
Data Processing:
- Integrate appropriate peak areas at each time point
- Convert absorbance to concentration using Beer-Lambert law and calibration curves
- Plot concentration versus time for all species of interest
- Differentiate concentration data to obtain instantaneous reaction rates
Kinetic Analysis:
- Plot reaction rate versus concentrations of potential rate-influencing species
- Test fits to various rate law models (zero-order, first-order, second-order)
- Determine reaction orders with respect to each component through mathematical modeling
Troubleshooting Notes:
- If spectral overlap complicates analysis, employ spectral deconvolution software
- For highly exothermic reactions, ensure adequate temperature control to maintain isothermal conditions
- Verify mixing efficiency to ensure homogeneous reaction conditions
Detailed Protocol: Reaction Progress NMR Kinetics
Purpose: To simultaneously monitor multiple species in a nucleophilic substitution reaction and identify potential intermediates using Nuclear Magnetic Resonance spectroscopy.
Materials and Equipment:
- NMR spectrometer with automated sampling capability or flow cell
- Deuterated solvents
- Internal standard (e.g., tetramethylsilane, 1,3,5-trimethoxybenzene)
- Temperature control unit
- Data processing software
Procedure:
- Method Development:
- Identify non-overlapping NMR signals for reactants, products, and potential intermediates
- Select appropriate internal standard that does not interfere with reaction
- Determine optimal pulse repetition time for quantitative measurements
Reaction Initiation:
- Prepare reaction mixture in NMR tube, including internal standard
- Insert into pre-equilibrated NMR spectrometer
- Commence automated data collection with predetermined time intervals
Data Collection:
- Acquire spectra at regular intervals throughout reaction progression
- Ensure sufficient signal-to-noise ratio while maintaining temporal resolution
- Continue until complete conversion or establishment of equilibrium
Data Processing:
- Integrate characteristic peaks for all species relative to internal standard
- Calculate concentrations using established response factors
- Generate concentration-time profiles for all components
Kinetic Analysis:
- Plot concentration trends to identify kinetic coupling between species
- Calculate rates from derivatives of smoothed concentration-time curves
- Test for consistency with proposed mechanisms
Applications in SN2X Research: Reaction progress NMR proves particularly valuable for distinguishing between classical SN2 and proposed SN2X mechanisms by potentially detecting stereochemical information and intermediates that would support a frontside attack pathway [35].
Analytical Framework for Distinguishing Substitution Mechanisms
Kinetic Signatures of Competing Mechanisms
The following diagram illustrates the diagnostic kinetic and analytical approaches for distinguishing between competing substitution mechanisms:
Quantitative Kinetic Parameters for Mechanism Discrimination
Table 2: Diagnostic Kinetic Criteria for Distinguishing Substitution Mechanisms
| Mechanistic Parameter | Classical SN2 | SN1 Mechanism | Proposed SN2X Mechanism |
|---|---|---|---|
| Rate Law Dependence | Second-order overall: Rate = k[Nu][Elec] [3] | First-order: Rate = k[Elec] | Potentially complex order; may show saturation kinetics |
| Steric Environment | Highly sensitive to steric hindrance; methyl > primary > secondary >> tertiary [3] | Accelerated by tertiary substitution | Potentially different steric requirements than SN2 |
| Stereochemical Outcome | Complete inversion of configuration [3] | Racemization (partial or complete) | Potential retention or partial inversion |
| Solvent Effects | Favored in polar aprotic solvents | Accelerated in polar protic solvents | Solvent dependence may differ from classical pathways |
| Leaving Group Effects | Rate dependent on leaving group ability | Rate dependent on leaving group ability | May show unusual leaving group dependence |
| Nucleophile Effects | Rate proportional to nucleophile strength and concentration [3] | Rate independent of nucleophile identity | May show unusual nucleophile structure-activity relationship |
| Catalyst Resting State | N/A (uncatalyzed) | N/A (uncatalyzed) | Could involve catalyst-substrate complex [35] |
| Diagnostic Experiments | Stereochemical tracing with chiral substrates [3] | Rate determination in different solvents | Kinetic isotope effects; advanced kinetic profiling |
Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for Kinetic Studies of Substitution Mechanisms
| Reagent/Material | Function in Kinetic Analysis | Application Examples | Technical Considerations |
|---|---|---|---|
| Deuterated Solvents | NMR spectroscopy for reaction monitoring; solvent effects studies [35] | Reaction progress NMR kinetics; solvent parameter correlation | Degree of deuteration; water content; chemical compatibility |
| Internal Standards | Quantitative reference for spectroscopic techniques [35] | Concentration determination in NMR, GC, HPLC | Chemical inertness; distinct spectroscopic signature |
| Anhydrous Salts | Controlling ionic strength; studying salt effects | Investigating ion pairing in substitution mechanisms | Purity; drying protocols; hygroscopicity |
| Isotopically Labeled Compounds | Kinetic isotope effect studies; mechanistic tracing | Distinguishing rate-determining steps; bond-breaking assessment | Isotopic purity; synthetic accessibility; cost |
| Chiral Substrates | Stereochemical outcome determination | Distinguishing inversion/retention in SN2X studies | Optical purity; configuration stability under conditions |
| Specialized Catalysts | Enabling novel reaction pathways | Investigating catalytic nucleophilic substitution | Air/moisture sensitivity; purification requirements |
| Stable Radical Inhibitors | Testing for radical mechanisms | Ruling out radical chain pathways in substitution | Redox compatibility; spectroscopic interference |
Advanced Applications in Pharmaceutical Research
For drug development professionals, kinetic analysis of substitution mechanisms provides critical insights for process optimization and impurity control. The application of RPKA to pharmaceutical reactions allows researchers to:
- Identify Rate-Determining Steps: Pinpoint the slowest step in complex reaction sequences to focus optimization efforts [35]
- Predict Impurity Formation: Understand how byproducts form through competing pathways to develop mitigation strategies
- Optimize Catalyst Systems: Determine optimal catalyst loading and identity through resting state analysis [35]
- Scale-up Prediction: Generate robust kinetic models that predict reaction behavior across scales
- Quality by Design (QbD): Establish design spaces for regulatory filings based on mechanistic understanding
Case studies in pharmaceutical development demonstrate the value of these approaches. For instance, kinetic analysis of Buchwald-Hartwig amination reactions resolved competing mechanistic proposals and enabled optimization of these important C-N bond-forming reactions in API synthesis [35]. Similarly, investigation of the organocatalyzed α-amination of aldehydes combined calorimetric and spectroscopic data to elucidate the complex reaction network and improve enantioselectivity [35].
For SN2X mechanism research specifically, the combination of advanced kinetic analysis with stereochemical and computational studies provides a powerful toolkit for establishing this non-classical pathway and developing its synthetic applications in medicinal chemistry.
Stereochemical Probes and Chiral Substrates for Mechanism Elucidation
Stereochemical probes and chiral substrates serve as powerful tools for elucidating reaction mechanisms, particularly in the study of nucleophilic substitution pathways. While the classic backside attack SN2 mechanism proceeds with complete inversion of configuration at chiral centers, recent research has uncovered more nuanced pathways such as the halogenophilic nucleophilic substitution (SN2X) mechanism, where the nucleophile approaches from the frontside relative to the leaving group [4] [36]. This technical guide examines the design and application of stereochemical probes within the context of frontside attack SN2X reaction mechanism research, providing researchers with both theoretical frameworks and practical methodologies for mechanistic investigation.
The fundamental principle underlying stereochemical probing relies on the distinct stereochemical outcomes associated with different reaction mechanisms. Whereas SN2 pathways typically proceed with inversion of configuration, and SN1 pathways with racemization, the emerging SN2X mechanism exhibits unique stereospecificity that can be distinguished through carefully designed chiral substrates [3] [36]. This guide details the synthesis, application, and analysis of such stereochemical probes, with particular emphasis on their role in characterizing unconventional nucleophilic substitution pathways relevant to pharmaceutical and synthetic chemistry.
SN2X Mechanism: Fundamentals and Stereochemical Implications
Distinguishing SN2X from Traditional Substitution Pathways
The halogenophilic nucleophilic substitution (SN2X) mechanism represents a significant departure from conventional substitution pathways. In traditional SN2 reactions, nucleophiles approach the carbon center from the side opposite the leaving group (backside attack), resulting in inversion of configuration at chiral centers [3]. In contrast, SN2X reactions feature nucleophilic attack directly toward the leaving group, often through a linear arrangement of the nucleophile, carbon center, and leaving group [4] [36].
This mechanistic distinction creates identifiable stereochemical signatures. Recent quantitative studies have developed parameters to characterize SN2X reactions, including the halogenophilic percentage (X%) and relative halogenophilicity (H), which correlate with established physical organic chemistry principles such as Hammett and Mayr postulates [4]. The SN2X pathway frequently coexists with traditional SN2 mechanisms, and their competition can be quantified through detailed kinetic analysis and stereochemical studies.
Table 1: Comparative Analysis of Nucleophilic Substitution Mechanisms
| Mechanism | Stereochemical Outcome | Nucleophile Approach | Key Identifying Features |
|---|---|---|---|
| SN2 | Inversion of configuration | Backside attack | Second-order kinetics, stereospecific inversion |
| SN1 | Racemization | Not applicable | First-order kinetics, carbocation intermediate |
| SN2X | Distinct stereospecificity | Frontside attack | Halogenophilic percentage, linear arrangement |
| Addition-Elimination | Retention or inversion | Varies | Five-coordinate intermediate, common at S(VI) |
Visualizing the SN2X Mechanism
Diagram 1: SN2X Reaction Pathway. This workflow illustrates the key steps in the halogenophilic substitution mechanism, highlighting the frontside attack and halogen bonding interactions.
The diagram above outlines the sequential process of the SN2X mechanism, beginning with chiral substrate recognition and proceeding through frontside nucleophile approach. The critical halogen bonding interaction between the nucleophile and leaving group distinguishes this pathway from conventional SN2 mechanisms [36]. This interaction facilitates an unusual linear arrangement in the transition state, ultimately leading to distinct stereochemical outcomes in the final product.
Stereochemical Probes for S(VI) Centers
Sulfur(VI) Functional Groups as Stereochemical Reporters
Sulfur(VI) centers provide particularly informative platforms for stereochemical mechanism studies due to their tetrahedral geometry and stability in various oxidation states. Key S(VI) functional groups employed as stereochemical probes include sulfoximines, sulfonimidamides, and sulfonimidoyl halides [37]. These scaffolds maintain configurational stability at sulfur while offering diverse reactivity profiles for nucleophilic substitution studies.
The resurgence of sulfur-fluoride exchange (SuFEx) chemistry, coined by Sharpless in 2014 as a 'click' reaction, has heightened interest in S(VI) stereochemistry [37] [38]. When substitution reactions occur at chiral S(VI) electrophiles, the stereochemical outcome provides direct mechanistic evidence. Four primary mechanisms have been proposed for nucleophilic substitution at aza-S(VI) systems: (1) SN1-type dissociation; (2) SN2-type inversion; (3) addition-elimination via a five-coordinate sulfurane intermediate; or (4) elimination-addition through a sulfene-type intermediate [37].
Table 2: Stereochemical Probes for S(VI) Center Mechanism Elucidation
| Chiral S(VI) Substrate | Leaving Group | Stereochemical Outcome | Proposed Mechanism |
|---|---|---|---|
| Sulfonimidoyl chlorides | Chloride | Inversion | SN2-type [37] |
| Sulfonimidoyl fluorides | Fluoride | Varies (inversion/retention) | Dependent on conditions [37] |
| Sulfonimidates | Alkoxy group | Inversion | SN2-type [37] |
| Sulfoximines | N/A | Retention (reduction) | Aluminum amalgam [37] |
Historical Development and Contemporary Applications
Seminal work by C. Johnson in 1971 established foundational principles for stereochemical studies at S(VI) centers. Johnson employed enantiopure sulfinamides as precursors to sulfonimidoyl chlorides, which were subsequently subjected to nucleophilic substitution with dimethylamine and sodium phenolate [37]. Through careful stereochemical tracing across multiple transformations, Johnson demonstrated that substitution at sulfonimidoyl chlorides proceeds with inversion of configuration, consistent with an SN2-type mechanism.
Contemporary applications of S(VI) stereochemical probes span medicinal chemistry and drug development. Chiral S(VI) centers in sulfoximines confer beneficial properties to drug-like compounds, including high solubility, polarity, and directional interactions with protein binding sites [37]. Pharmaceutical agents incorporating chiral S(VI) centers, such as anti-inflammatory DFV890 and anticancer compounds VIP152 and ceralasertib, have advanced to Phase II and Phase III clinical trials, highlighting the practical significance of stereochemical control at sulfur centers [37].
Experimental Protocols for Stereochemical Mechanism Studies
Synthesis of Enantioenriched Sulfonimidoyl Chlorides
Principle: Electrophilic chlorination of enantiopure sulfinamides generates configurationally stable sulfonimidoyl chlorides that serve as versatile stereochemical probes [37].
Detailed Protocol:
- Begin with enantiopure sulfinamide (e.g., S-1, 1.0 equiv.) dissolved in anhydrous dichloromethane (0.1 M concentration) under inert atmosphere.
- Cool the solution to -78°C using a dry ice/acetone bath.
- Add chlorinating agent (e.g., tert-butyl hypochlorite, 1.1 equiv.) dropwise with stirring.
- Maintain reaction at -78°C for 30 minutes, then warm gradually to room temperature over 2 hours.
- Monitor reaction completion by TLC or NMR spectroscopy.
- The resulting sulfonimidoyl chloride (e.g., R-2) may be used directly or purified by flash chromatography under anhydrous conditions.
Stereochemical Analysis: The chlorination step proceeds with retention of configuration at sulfur, as electrophilic substitution occurs without perturbation of the tetrahedral structure [37]. This conservation of stereochemistry enables subsequent nucleophilic substitution studies with defined initial configuration.
Nucleophilic Substitution with Stereochemical Tracing
Principle: Treatment of enantioenriched sulfonimidoyl chlorides with nucleophiles reveals mechanistic pathway through stereochemical analysis of products.
Detailed Protocol:
- Prepare sulfonimidoyl chloride (e.g., R-2, 1.0 equiv.) in anhydrous tetrahydrofuran (0.1 M).
- Add nucleophile (e.g., dimethylamine, 2.0 equiv.) dropwise at 0°C with vigorous stirring.
- Warm reaction to room temperature and monitor by TLC until complete.
- Quench with saturated aqueous ammonium chloride and extract with ethyl acetate (3 × 20 mL).
- Combine organic layers, dry over anhydrous magnesium sulfate, and concentrate under reduced pressure.
- Purify the resulting sulfonimidamide (e.g., R-3) by flash chromatography.
Stereochemical Analysis:
- Convert sulfonimidamide product to sulfinamide via reduction with aluminium amalgam.
- Compare specific rotation of resulting sulfinamide with authentic samples.
- Alternatively, employ chiral HPLC or X-ray crystallography for direct stereochemical assignment.
Key Finding: Nucleophilic substitution at sulfonimidoyl chlorides proceeds with inversion of configuration, consistent with an SN2-type mechanism [37].
Synergistic Hydrogen Bonding Phase-Transfer Catalysis
Principle: Combined chiral hydrogen bond donor and onium salt catalysts enable enantioconvergent substitutions through ternary complex formation [39].
Detailed Protocol:
- Charge racemic benzylic bromide (e.g., rac-1a, 1.0 equiv.), KF (2.5 equiv.), and molecular sieves (4Å) to flame-dried flask.
- Add catalysts: chiral bis-urea HBD (e.g., (S)-3h, 10 mol%) and tetraarylphosphonium iodide (e.g., Ph₄P⁺I⁻, 10 mol%).
- Evacuate and backfill with argon (3 cycles).
- Add dry p-xylene (0.25 M concentration relative to substrate) via syringe.
- Stir reaction at 15°C until complete conversion (monitored by TLC or GC/MS).
- Filter through Celite, concentrate under reduced pressure, and purify by flash chromatography.
Mechanistic Insight: The chiral bis-urea catalyst and onium salt form a well-characterized ternary HBD–onium fluoride complex that enables enantiodiscrimination of racemic electrophiles [39]. Diffusion-ordered NMR spectroscopy (DOSY) confirms the molecular weight of the catalyst-resting state corresponds to a 1:1 catalyst–substrate complex.
Analytical Methods for Stereochemical Determination
Comparative Methodologies for Configuration Assignment
The evolution of analytical technologies has transformed stereochemical analysis from reliance on optical rotation measurements to more definitive techniques including chiral HPLC and X-ray crystallography [37]. Contemporary mechanistic studies employ multiple orthogonal methods to unambiguously assign stereochemical outcomes.
Table 3: Analytical Techniques for Stereochemical Analysis
| Method | Application | Key Information | Limitations |
|---|---|---|---|
| Optical Rotation | Historical configuration comparison | Relative configuration changes | Requires authentic standards, less reliable |
| Chiral HPLC | Direct enantiopurity assessment | Enantiomeric ratio, absolute configuration | May require derivatization |
| X-ray Crystallography | Definitive configuration assignment | Absolute stereochemistry, molecular geometry | Requires suitable crystals |
| NMR Spectroscopy | Structural characterization | Diastereotopic proton analysis, DOSY for complexation | Limited for enantiomer distinction |
| Kinetic Analysis | Mechanistic pathway elucidation | Reaction rates, activation parameters | Indirect stereochemical evidence |
Workflow for Comprehensive Stereochemical Analysis
Diagram 2: Stereochemical Analysis Workflow. This pathway outlines the sequential process for comprehensive stereochemical analysis, from substrate preparation to mechanistic interpretation.
The analytical workflow begins with preparation of enantiopured substrates, proceeds through substitution and purification, and employs multiple characterization techniques to unambiguously assign configuration. Correlation of stereochemical outcome with reaction conditions enables mechanistic interpretation, distinguishing between SN2, SN2X, and other pathways.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Stereochemical Mechanism Studies
| Reagent/Catalyst | Function | Application Example | Mechanistic Role |
|---|---|---|---|
| Chiral bis-urea HBD | Hydrogen bond donor catalyst | Enantioconvergent fluorination [39] | Forms ternary complex with onium fluoride |
| Tetraarylphosphonium salts | Phase-transfer co-catalyst | Solubilizing KF in organic media [39] | Enhances fluoride nucleophilicity |
| Sulfonimidoyl chlorides | Chiral electrophilic probes | Stereochemical outcome studies [37] | SN2-type inversion demonstration |
| Aluminium amalgam | Reducing agent | Sulfoximine to sulfinamide reduction [37] | Stereospecific reduction with retention |
| Enantiopure sulfinamides | Chiral precursors | Sulfonimidoyl chloride synthesis [37] | Configurationally defined starting materials |
Stereochemical probes and chiral substrates provide indispensable tools for elucidating complex nucleophilic substitution mechanisms, including unconventional pathways like the SN2X reaction. Through careful design of stereochemical experiments, synthetic chemists can distinguish between competing mechanistic pathways based on distinct stereochemical outcomes. The protocols and methodologies outlined in this technical guide equip researchers with robust frameworks for investigating frontside attack mechanisms, with particular relevance to pharmaceutical development and advanced synthetic methodology. As stereochemical probing techniques continue to evolve alongside analytical technologies, our understanding of nucleophilic substitution mechanisms will undoubtedly deepen, enabling new strategic approaches to asymmetric synthesis.
Within the broad landscape of chemical reactivity, the bimolecular nucleophilic substitution (SN2) reaction serves as a fundamental prototype for understanding reaction mechanisms [40]. The canonical mechanism involves a concerted backside attack, leading to inversion of stereochemistry [3]. However, the research frontier extends to exploring variations such as the frontside attack mechanism (often denoted in broader studies as SN2X or within the continuum of SN2/SH2 mechanisms), which presents a distinct electronic and steric landscape on the potential energy surface (PES) [9]. Computational chemistry is indispensable for modeling these reaction pathways, particularly for locating and characterizing transition states (TS)—the fleeting, high-energy saddle points that dictate reaction kinetics and selectivity [41]. This guide details the core methodologies for modeling transition states and reaction pathways, contextualized within advanced SN2 reaction mechanism research.
Foundational Theory: The Potential Energy Surface of SN2 Reactions
The SN2 reaction profile is classically described by a double-well PES, featuring a reactant complex (RC), a central transition state (TS), and a product complex (PC) before dissociation to final products [9] [40]. A striking discovery is that changing the central atom from carbon (second-period) to silicon or germanium (higher-period) can transform this double-well PES into a single-well PES, indicative of a stable pentavalent intermediate [9]. Conversely, manipulating nucleophilicity, leaving group ability, and steric bulk can revert a single-well back to a double-well or even create a single-well for carbon [9]. This fluidity underscores the necessity for accurate TS modeling to predict and rationalize reactivity.
Table 1: Key Features of SN2 Reaction Potential Energy Surfaces
| Feature | Double-Well PES (e.g., Cl⁻ + CH₃Cl) | Single-Well PES (e.g., SN2@Si) | Implications for TS Search |
|---|---|---|---|
| Stationary Points | Reactants → RC → TS → PC → Products | Reactants → Stable Intermediate → Products | TS may be lower in energy than separated reactants (submerged barrier). |
| Central Barrier | Positive ΔE‡,centr (TS relative to RC) [9]. | May be absent or very low. | Locating the TS requires methods sensitive to shallow or submerged barriers. |
| Solvent Effect | Polar solvents stabilize ions, raising barrier, can change to unimodal PES [9]. | Large LUMO central atoms (Si, As) show more gradual charge transfer, resisting solvent-induced PES changes [9]. | Solvation models are critical for realistic modeling; gas-phase PES differs drastically. |
| Dynamical Bottleneck | The TS remains a dynamical bottleneck even if energy is submerged [40]. | The stable intermediate well may control kinetics. | Identifying the rate-determining step is key; may not be a traditional TS. |
Methodologies for Transition State Location and Validation
Locating a first-order saddle point on a high-dimensional PES is non-trivial. The following are key methodological approaches.
Synchronous Transit and Interpolation Methods
These methods use interpolated geometries between reactants and products as a starting guess for the reaction path.
- Linear Synchronous Transit (LST): A naïve linear interpolation in internal or Cartesian coordinates. It often yields poor, high-energy guesses with unphysical geometries (e.g., atoms passing through each other) [42].
- Quadratic Synchronous Transit (QST): Uses quadratic interpolation for a more flexible constraining curve. QST3 is a robust variant that requires input of reactant, product, and an initial TS guess. It iteratively finds the maximum along the curve and optimizes the geometry normal to it until convergence [42].
- Experimental Protocol for QST3:
- Geometry Optimization: Fully optimize the structures of the reactant (R) and product (P) at an appropriate level of theory (e.g., DFT/B3LYP/6-31+G*).
- Generate TS Guess: Create an initial guess, often via linear interpolation or chemical intuition. For a frontside SN2 attack, this may involve positioning the nucleophile near the leaving group on the same side of the central carbon.
- QST3 Calculation: Submit a calculation specifying the three structures (R, P, TS-guess). The algorithm will interpolate, find a maximum, and optimize.
- Verification: Confirm the output has exactly one imaginary vibrational frequency corresponding to the expected bond formation/breaking motion.
- Experimental Protocol for QST3:
Nudged Elastic Band (NEB) and String Methods
These are "double-ended" methods that refine an entire path between endpoints.
- Nudged Elastic Band (NEB): Multiple "images" of the system are placed along an initial guess path (e.g., from interpolation). Images are connected by spring forces and optimized, but only the component of the true gradient perpendicular to the path is used to "nudge" images downhill. This prevents corner-cutting and converges to an approximation of the minimum energy path (MEP) [42].
- Climbing-Image NEB (CI-NEB): An enhancement where the highest-energy image is not affected by springs and is allowed to "climb" upwards along the path tangent while being minimized in perpendicular directions. This image converges directly to the TS [42].
- Freezing String Method (FSM): A modern double-ended method. Strings of geometries from reactants and products "grow" towards each other within the reaction channel until they connect. The peak along the connected path serves as an excellent TS guess and can be optimized rapidly, especially when paired with neural network potentials (NNPs) [43].
- Experimental Protocol for CI-NEB:
- Prepare Endpoints: Optimize R and P structures.
- Generate Initial Path: Create 5-15 intermediate images, typically via linear interpolation.
- CI-NEB Setup: Specify the band of images, a spring constant, and a climbing image criterion. The calculation will simultaneously optimize all images.
- Analysis: The climbing image's geometry is the TS guess. Its single imaginary frequency and an intrinsic reaction coordinate (IRC) calculation confirm it connects to your R and P.
- Experimental Protocol for CI-NEB:
Machine Learning and Neural Network Potentials (NNPs)
These are revolutionizing the speed of PES exploration.
- Neural Network Potentials (e.g., ANI-2x, AIMNet2): Trained on DFT data, NNPs provide energies and forces orders of magnitude faster than ab initio methods. They enable rapid sampling of conformational space and reaction pathways, making thorough TS searches and IRC calculations feasible for larger systems [44] [43].
- Direct TS Prediction Models (e.g., React-OT): End-to-end machine learning models predict TS structures directly from reactants and products. React-OT starts from a linear interpolation guess and refines it in ~5 steps (~0.4 seconds), achieving high accuracy [41]. This is transformative for high-throughput screening.
Table 2: Comparison of Computational Methods for TS Location
| Method | Type | Required Input | Computational Cost | Strengths | Weaknesses |
|---|---|---|---|---|---|
| QST3 | Interpolation | R, P, TS Guess | Low-Moderate (DFT) | Robust, good with a plausible guess [42]. | Requires a TS guess; struggles with multi-step paths. |
| CI-NEB | Double-ended Path | R, P, Initial Path (Images) | High (DFT), Low (NNP) | Finds MEP and TS directly; handles intermediates. | Cost scales with number of images; needs careful setup. |
| Freezing String Method (FSM) | Double-ended Path | R, P | Moderate (DFT), Very Low (NNP) | Robust to internal rotations; fast connection [43]. | Emerging method; integration varies by software. |
| React-OT (ML) | Direct Prediction | R, P | Very Low | Extremely fast; high accuracy for trained chemistries [41]. | Dependent on training data domain; black-box nature. |
| Dimer Method | Single-ended | Initial Geometry | Moderate | Doesn't require R & P; follows low-curvature path [42]. | Can get lost in complex PES; common in solid-state. |
Visualization of Workflows and Pathways
Title: Computational Workflow for SN2 Transition State Search
Title: Comparison of SN2 Potential Energy Surface Types
Table 3: Key Research Reagent Solutions for Computational SN2 Studies
| Item/Resource | Function & Relevance | Example/Note |
|---|---|---|
| Quantum Chemistry Software | Performs electronic structure calculations (DFT, ab initio) for energy, gradient, and Hessian evaluations. | Gaussian, ORCA, Q-Chem, PySCF. Essential for final TS validation and benchmarking. |
| Neural Network Potential (NNP) Platforms | Provides ultra-fast, approximate energies/forces for rapid sampling and preliminary TS searches [44] [43]. | ANI-2x, AIMNet2, CHGNET. Integrated into packages like Rowan, TorchANI. |
| TS Search & Pathfinder Algorithms | Implements algorithms like QST, NEB, Dimer, FSM to locate saddle points. | Implemented in major software (Gaussian, ASE, LAMMPS) or specialized packages (ML-FSM) [43]. |
| Solvation Model Parameters | Models solvent effects (polar, apolar) critical for realistic SN2 barriers, as gas-phase and solution PES differ drastically [9]. | PCM, SMD, COSMO. Parameters for water, DMSO, THF, etc. |
| Benchmark Reaction Datasets | Curated sets of known reactions with validated TS structures for training ML models and benchmarking methods. | Data from studies like [9] [41]. The 9,000-reaction set used to train React-OT is an example [41]. |
| Kinetic Analysis Tools | Extracts rate constants from computed energies (e.g., via Transition State Theory). | Can involve RRKM/master equation calculations for gas-phase dynamics [40]. |
| Visualization & Analysis Software | Analyzes geometries, vibrational modes, IRC paths, and charge distribution. | VMD, Jmol, Multiwfn, IBOView. Critical for diagnosing TS and understanding electronic shifts [9]. |
Advanced Spectroscopic Techniques for Reaction Intermediate Characterization
Elucidating the precise sequence of elementary steps in a chemical reaction is a fundamental pursuit in physical organic chemistry and catalysis. The identification and characterization of reactive intermediates—transient species with lifetimes ranging from femtoseconds to seconds—provide the most direct evidence for a proposed mechanism. This endeavor is particularly challenging in the study of novel substitution pathways, such as the hypothesized frontside attack nucleophilic substitution (SN2X) mechanism. In contrast to the well-established backside attack SN2 mechanism, which proceeds with a characteristic inversion of stereochemistry at a saturated carbon center [3] [1], a frontside attack would imply retention of configuration. While computational studies may propose such pathways, experimental validation hinges on the ability to capture and analyze the short-lived intermediates that define this alternative route [1]. This technical guide surveys advanced spectroscopic techniques that have revolutionized our capacity to interrogate these elusive species, focusing on their principles, applications, and implementation for researchers investigating complex reaction mechanisms like the SN2X.
Online Mass Spectrometry (Real-time MS)
Online or real-time mass spectrometry has emerged as a powerful tool for monitoring catalytic cycles and capturing intermediates as they form in solution. Its principal advantage is the continuous, temporally resolved delivery of reaction mixture data, providing a dynamic view of interconversions rather than static snapshots [45].
Principle and Application: Electrospray Ionization (ESI) gently transfers solution-phase ions into the gas phase for mass analysis. When coupled directly to a reaction vessel via a flow system, it allows for real-time detection. This technique is exceptionally sensitive to low-abundance charged species and is ideal for studying organometallic, photocatalytic, and enzymatic reactions where intermediates may be intrinsically ionic or can be "charge-tagged" [46]. A landmark study demonstrated its power by capturing multiple radical and resonance-stabilized intermediates in the P450-catalyzed oxidative dimerization of 1-methoxynaphthalene, elucidating the complete catalytic cycle [45].
Detailed Experimental Protocol (Online ESI-MS for Enzymatic Intermediates):
- Reaction Setup: Prepare the enzymatic reaction in a buffer compatible with ESI (e.g., 500 mM ammonium acetate, pH 7.5) to maintain enzyme stability and efficient ionization [45].
- Continuous Sampling: Use a custom-built or commercial microfluidic/pressurized infusion setup. A mixing tee continuously dilutes the ongoing reaction mixture with a compatible solvent to quench secondary reactions and stabilize intermediates within the generated microdroplets [45].
- Real-time Ionization and Detection: The diluted stream is directly infused into an ESI source operating at typical conditions (e.g., +5 kV spray voltage, 110 psi nebulizing gas). The high-resolution mass spectrometer (e.g., Q-TOF, Orbitrap) acquires spectra continuously from reaction initiation.
- Data Analysis: Extract ion chromatograms for masses corresponding to postulated intermediates and products. Temporal profiling of their relative abundances maps out the reaction pathway. Tandem MS (MS/MS) on mass-selected ions provides structural validation [45] [46].
Online ESI-MS Workflow for Intermediate Capture
Time-Resolved X-ray and Optical Spectroscopies
For intermediates with distinct optical or structural signatures, time-resolved methods offer direct, element-specific insight.
Time-Resolved X-ray Free-Electron Laser (XFEL) Crystallography: This technique uses ultra-bright, femtosecond X-ray pulses to capture molecular structures at near-atomic resolution before radiation damage occurs. It can "film" enzymatic reactions by triggering catalysis within a crystal and then probing it at defined time delays. It has been used to capture nitric oxide-bound and radical intermediates in P450nor enzymatic reactions [45].
Rapid-Scan and Stopped-Flow Spectroscopies (UV-Vis, IR, Raman): These methods mix reactants rapidly and monitor spectral changes on millisecond to second timescales. They are excellent for tracking intermediates with characteristic absorbances (e.g., metal-oxo species in Compound I of P450s) or vibrational frequencies [45]. Multidimensional techniques like time-resolved resonance Raman can provide detailed structural information about chromophoric centers.
Advanced Mass Spectrometric Structural Elucidation
Beyond mere mass detection, tandem MS techniques provide critical structural information on detected intermediates.
Collision-Induced Dissociation (CID) and Energy-Dependent Fragmentation: By fragmenting mass-selected ions, CID patterns can distinguish between structural isomers, such as a reactive Pd(IV) intermediate and its isobaric Pd(II) product complex, based on different fragmentation pathways [46]. Quantitative analysis of energy-dependent fragmentation yields can even provide bond dissociation energies for intermediates, offering thermodynamic insights [46].
Ion Mobility-Mass Spectrometry (IM-MS): IM-MS separates ions based on their size and shape (collisional cross-section) in addition to mass. This is invaluable for identifying intermediates that are structural isomers or conformers of other species with identical mass, a common challenge in organometallic mechanisms [46].
Nuclear Magnetic Resonance (NMR) and Electron Paramagnetic Resonance (EPR) Spectroscopy
Time-Resolved NMR: Though traditionally slower, advanced NMR methods like rapid mixing and flow techniques can detect unstable intermediates on the seconds-to-minutes timescale. It provides unparalleled atomic-level detail about connectivity and environment, as demonstrated in studies of acetyl-CoA synthetase [45].
EPR Spectroscopy: This is the definitive technique for detecting and characterizing paramagnetic intermediates, such as organic radicals or metal centers with unpaired electrons, which are often proposed in radical-based mechanisms like those in certain P450 cycles or radical SN2-type pathways [45].
Quantitative Comparison of Key Techniques
Table: Comparison of Advanced Spectroscopic Techniques for Intermediate Characterization
| Technique | Time Resolution | Primary Information Gained | Key Strength | Major Limitation | Ideal for SN2X Studies? |
|---|---|---|---|---|---|
| Online ESI-MS | Seconds | Mass, temporal abundance, structure via MSⁿ | Sensitive, real-time monitoring of multiple species, works in solution. | Requires ionizable species; may generate gas-phase artifacts. | High, for charged/charge-tagged intermediates. |
| Time-Resolved XFEL | Femtoseconds | Atomic-resolution 3D structure. | "Direct observation" of geometry changes in crystals. | Requires crystallizable systems; not for homogeneous solution. | Low, unless transition state analogs are crystallized. |
| Stopped-Flow UV-Vis/IR | Milliseconds | Electronic/Vibrational structure. | Excellent for chromophoric intermediates (e.g., metal-oxo). | Requires distinct optical signature; less specific. | Medium, if intermediates have unique absorbance. |
| CID/IM-MS | N/A (Post-ionization) | Fragmentation patterns, ion size/shape. | Distinguishes isobaric/isomeric intermediates. | Requires successful prior MS detection. | High, for structural validation of MS-detected ions. |
| Time-Resolved NMR | Seconds to Minutes | Atomic connectivity, chemical environment. | Ultimate structural detail in solution. | Low sensitivity; requires relatively long-lived species. | Low, due to likely short lifetimes. |
| EPR Spectroscopy | Microseconds to Seconds | Identification of paramagnetic centers, spin density. | Definitive for radical intermediates. | Only for paramagnetic species. | High, if radical pairs or metal-centered radicals are involved. |
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents and Materials for Intermediate Trapping Studies
| Item | Function/Description | Example from Literature |
|---|---|---|
| Charge-Tagging Reagents | Incorporate a permanent ionic group (e.g., ammonium, phosphonium) into a substrate or catalyst ligand to ensure efficient MS detection of otherwise neutral intermediates [46]. | Pyridinium-tagged directing groups in Pd-catalyzed C-H activation studies. |
| Radical Traps (Spin Traps) | React selectively and rapidly with radical intermediates to form stable, detectable adducts. Used with EPR or MS. | TEMPO (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl), used to trap and identify radical intermediates in P450 catalysis for MS analysis [45]. |
| Rapid-Mixing Devices | Ensure sub-second mixing of reactants to initiate synchronous catalysis for time-resolved studies. | Stopped-flow apparatus, continuous-flow microfluidic chips. |
| MS-Compatible Buffers | Volatile salts that maintain pH and protein stability without suppressing ionization or causing source contamination. | Ammonium acetate, ammonium bicarbonate. Used at high concentration (e.g., 500 mM) to stabilize enzymes for online MS [45]. |
| Ultrafast Triggering Sources | Initiate reactions photochemically or electrochemically on ultrafast timescales for pump-probe experiments. | Femtosecond laser pulses (for photolysis), pulsed electrodes. |
Decision Flow for Intermediate Characterization Technique
The quest to characterize reactive intermediates drives the development of ever-more sophisticated spectroscopic and spectrometric techniques. For probing contentious mechanisms like the frontside attack SN2X pathway, a multi-technique approach is essential. Real-time MS offers the best chance to capture fleeting species directly from solution, while advanced tandem MS methods can validate their structures. Correlative evidence from time-resolved optical or X-ray techniques on systems designed to slow down key steps can provide complementary geometric and electronic information. The integration of these tools, guided by mechanistic hypotheses and aided by strategic reagent design (e.g., charge tags, radical traps), empowers researchers to move beyond indirect kinetic evidence and directly observe the molecular actors in chemical transformations, thereby definitively testing the boundaries of established mechanistic paradigms like the SN2 backside attack rule [3] [1].
Isotope Labeling Studies in Mechanistic Investigation
Isotope labeling is a foundational technique in scientific research that leverages the unique physical properties of isotopes to trace molecular dynamics in biological, chemical, and physical processes. By replacing specific atoms in a compound with their isotopic counterparts, researchers gain unparalleled insights into reaction pathways, metabolic fluxes, and mechanistic details [47]. This approach has a long tradition in biosynthesis studies and has experienced a revival in recent years, particularly as genome sequencing technologies have enabled rapid access to biosynthetic genes and enzymes [48]. In the specific context of investigating nucleophilic substitution mechanisms, particularly the proposed frontside attack SN2X reaction mechanism, isotope labeling provides critical experimental evidence that cannot be obtained through other analytical methods.
The fundamental principle underlying isotope labeling rests on the chemical equivalence of isotopes—they share identical electronic configurations and reactivity—while offering physical detectability through either radioactive decay or mass differences [47]. This dual nature allows researchers to "tag" molecules without altering their chemical behavior, then track these tagged molecules through complex reaction mechanisms using appropriate detection methods. For mechanistic studies in organic chemistry, this capability proves invaluable for distinguishing between alternative reaction pathways, identifying rate-determining steps, and elucidating stereochemical outcomes.
Fundamental Principles of Isotope Labeling
Types of Isotopes and Their Properties
Isotope labeling strategies employ both radioactive and stable isotopes, each with distinct advantages for mechanistic investigations. The selection of appropriate isotopes depends on the specific research question, detection capabilities, and safety considerations [47] [49].
Table: Key Isotopes and Their Applications in Mechanistic Studies
| Isotope | Type | Primary Applications | Detection Methods |
|---|---|---|---|
| ³H | Radioactive | Tracking reaction sites in organic molecules | Scintillation counting, autoradiography |
| ¹⁴C | Radioactive | Metabolic pathway tracing, reaction mechanism studies | Scintillation counting, AMS |
| ³²P | Radioactive | Phosphorylation studies, nucleic acid tracking | Autoradiography, scintillation counting |
| ¹³C | Stable | Metabolic flux analysis, reaction mechanism elucidation | NMR, MS |
| ¹⁵N | Stable | Protein structural studies, nitrogen metabolism | NMR, MS |
| ¹⁸O | Stable | Oxygen source tracing in oxidation reactions | MS |
| ²H | Stable | Reaction pathway tracing, kinetic isotope effects | NMR, MS |
Radioactive isotopes, such as ³H, ¹⁴C, and ³²P, emit detectable radiation (e.g., β-particles) and are typically tracked via scintillation counters or autoradiography [47]. These isotopes offer exceptional sensitivity, with detection limits as low as 10⁻¹⁴ to 10⁻¹⁸ grams, surpassing traditional analytical methods by several orders of magnitude [49]. However, their use requires specialized safety protocols and facilities, which may limit application in some research environments.
Stable isotopes, including ¹³C, ¹⁵N, ¹⁸O, and ²H (deuterium), are non-radioactive and analyzed by mass spectrometry (MS) based on mass shifts or by nuclear magnetic resonance (NMR) spectroscopy based on isotopic displacement [47] [49]. These isotopes have gained prominence in mechanistic studies due to their safety profile and compatibility with advanced analytical techniques like LC-MS/MS and high-field NMR. The absence of radiation hazards facilitates experimental procedures and allows for longer-term studies without radioactive decay limitations.
Basic Labeling Strategies
Isotope labeling approaches can be categorized into several strategic frameworks based on experimental design:
Single Labeling: This approach targets one specific atom position within a compound (e.g., [¹³C]-glucose) to track the fate of that particular atom through a reaction mechanism [47]. In the context of SN2X mechanism studies, single labeling at the reaction center can reveal whether bond formation and breakage occur simultaneously or through discrete steps.
Parallel Labeling: This method uses multiple isotopes simultaneously (e.g., [¹³C]-glucose + [²H]-water) to reduce biological variability and enhance data robustness [47]. For complex mechanistic questions, parallel labeling can provide complementary information about different aspects of the reaction pathway.
Isotope Dilution Methods: These techniques use isotopically labeled internal standards for absolute quantification [50]. Stable-isotope dilution (SID) methodology in combination with LC-MS/MS provides the highest possible analytical specificity for quantitative determinations [51]. This approach is particularly valuable for accurately measuring intermediate concentrations in kinetic studies of reaction mechanisms.
Dual Labeling: This strategy incorporates two different isotopes within the same molecule to track multiple positions simultaneously, providing information about molecular fragmentation and recombination patterns during chemical reactions.
Isotope Labeling Techniques and Methodologies
Experimental Workflows for Mechanistic Studies
The application of isotope labeling to mechanistic investigation follows systematic experimental workflows that vary based on the isotopic label and detection method employed.
Table: Comparison of Isotope Labeling Methodologies
| Methodology | Isotopes Used | Key Steps | Applications in Mechanism Studies |
|---|---|---|---|
| Radioactive Isotope Labeling | ³H, ¹⁴C, ³²P, ³⁵S | 1. Label selection 2. Sample incorporation 3. Detection via autoradiography or scintillation counting | Tracking atom positions in product formation, determining kinetic parameters |
| Stable Isotope Metabolic Labeling | ¹³C, ¹⁵N, ²H | 1. Introduction of labeled substrates 2. Metabolic incorporation 3. MS or NMR analysis | Elucidating reaction pathways, quantifying flux through alternative mechanisms |
| SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture) | ¹³C/¹⁵N-arginine/lysine | 1. "Heavy" media preparation 2. Cell culture incorporation 3. Quantitative proteomics via MS | Studying enzyme expression and turnover in mechanistic contexts |
| Isotope-Coded Affinity Tags (ICAT) | ²H/¹³C | 1. Chemical tagging of functional groups 2. Affinity purification 3. MS analysis | Identifying reactive intermediates in complex reaction mixtures |
For radioactive isotope labeling, the standard workflow begins with careful selection of an appropriate isotope matched to the target molecules (e.g., ¹⁴C for carbon reaction centers) [47]. The labeled compound is then introduced into the reaction system, whether in cell-free chemical systems, cell cultures, or animal models. After the reaction proceeds, detection typically involves autoradiography or liquid scintillation counting to track the position and quantity of the radioactive label in products and intermediates.
Stable isotope labeling approaches, particularly those using ¹³C, have revolutionized mechanistic studies in complex systems [52]. Metabolic Flux Analysis (¹³C-MFA) utilizes ¹³C-labeled substrates to trace carbon flow through reaction networks, with isotopomer analysis via MS providing data on metabolic intermediates that can be used to model flux networks [47]. This approach is especially powerful for studying enzymatic mechanisms in their native biological contexts.
Advanced Labeling Strategies
Recent technological advances have spawned sophisticated labeling strategies that enhance the precision and scope of mechanistic investigations:
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC): This technique involves culturing cells in media containing isotopically labeled amino acids (e.g., ¹³C/¹⁵N-arginine/lysine) [47] [53]. The "heavy" amino acids are incorporated into proteins during synthesis, allowing precise quantification and comparison of protein expression levels between different experimental conditions using mass spectrometry.
Tandem Mass Tags (TMT) and Isobaric Tagging: These multiplexing approaches use isotope-encoded reagents that covalently bind to peptides or other molecules [53]. All versions of each tag have the same molecular mass, but the positions of heavy and light isotopes are adjusted to affect the mass of a "reporter ion" region upon fragmentation in MS/MS. This enables simultaneous comparison of multiple samples in a single MS run, significantly enhancing throughput and reducing quantitative variability.
Isotope Dilution Mass Spectrometry (IDMS): This reference technique for quantitative analysis combines the sensitivity and selectivity of MS instruments with the precision and accuracy associated with the use of internal standards [50]. Isotope-labeled analogs serve as ideal internal standards because they closely mimic the behavior of their natural counterparts during analytical processing while being distinguishable by MS.
Diagram 1: Generalized workflow for isotope labeling studies in mechanistic investigation, showing the key stages from experimental design to mechanistic validation.
Application to Nucleophilic Substitution Mechanisms
SN2 Reaction Mechanism Fundamentals
The SN2 (substitution nucleophilic bimolecular) reaction represents one of the most fundamental processes in organic chemistry, characterized by a concerted mechanism in which bond formation between the nucleophile and electrophilic carbon occurs simultaneously with bond cleavage between the electrophilic carbon and the leaving group [54] [3]. This reaction follows second-order kinetics, with the rate dependent on both the nucleophile and substrate concentrations [54]. A defining feature of the classic SN2 mechanism is its stereochemical outcome—the reaction proceeds exclusively via backside attack, resulting in inversion of configuration at the reaction center [3] [1].
The backside attack mechanism arises from electronic considerations: the nucleophile donates a pair of electrons into the most accessible empty orbital, which is the antibonding (σ*) orbital of the carbon-leaving group bond, residing at 180° to the bond [3]. This donation results in cleavage of the bond as the new bond forms. The requirement for backside attack stems from repulsive interactions that would occur if the nucleophile approached from the same side as the leaving group, where like charges would repel each other [1]. The transition state for this concerted process features trigonal bipyramidal geometry at carbon, with partial bonds to both the nucleophile and leaving group [3].
Investigating the SN2X Mechanism via Isotope Labeling
The proposed frontside attack SN2X mechanism represents a significant departure from the classical SN2 pathway and requires sophisticated experimental approaches for verification. Isotope labeling studies provide critical tools for distinguishing between these mechanistic possibilities through several strategic applications:
Kinetic Isotope Effects (KIEs): KIEs modulate reaction rates depending on the isotopic composition of substrate(s) and can provide definitive evidence about rate-determining steps and transition state geometry [52]. Primary KIEs, which occur when a bond to the isotopic atom is broken or formed in the rate-determining step, can be particularly informative. For instance, comparing reaction rates with ¹²C vs. ¹³C at the reaction center can reveal whether rehybridization occurs during the transition state. Secondary KIEs, which occur when the isotopic atom is not directly involved in bond cleavage/formation but is adjacent to the reaction center, can provide information about changes in steric congestion or hyperconjugation during the transition state.
Isotopic Tracing of Reaction Pathways: By strategically placing isotopic labels at specific atomic positions in reactants and analyzing their positions in products, researchers can track molecular reorganization during the reaction [48] [47]. For the SN2X mechanism, dual labeling experiments could determine whether the proposed frontside attack leads to different atom mapping in products compared to the classical backside attack mechanism.
Stereochemical Analysis Using Isotopic Probes: Isotope labeling enables precise determination of stereochemical outcomes when combined with chiral analysis techniques. For instance, using stereospecifically deuterated substrates in combination with ¹H or ²H NMR can reveal whether inversion, retention, or racemization occurs at the reaction center [1]. This approach provides direct experimental evidence for distinguishing between frontside and backside attack mechanisms.
Diagram 2: Conceptual framework for distinguishing classical SN2 and SN2X mechanisms using isotope labeling approaches, highlighting key stereochemical and kinetic differentiators.
Kinetic Isotope Effects in Mechanism Elucidation
Kinetic isotope effects (KIEs) represent one of the most powerful applications of isotope labeling in mechanistic studies, providing intimate information about transition state structure [52]. KIEs arise because bonds to heavier isotopes (e.g., ¹³C vs. ¹²C, ²H vs. ¹H) have lower zero-point vibrational energies and thus require more energy to break. When a bond to the isotopically labeled atom is broken or formed in the rate-determining step, a primary KIE is observed, typically with kₗᵢgₕₜ/kₕₑₐᵥᵧ values ranging from 1.02-1.04 for ¹³C/¹²C and 2-7 for ²H/¹H.
For the SN2X mechanism investigation, KIEs can provide critical evidence:
Primary ¹⁴C KIEs: When the isotopic label is placed at the reaction center carbon, classical SN2 reactions typically exhibit normal KIEs (k₁₂C/k₁₄C > 1) due to rehybridization from sp³ to sp²-like character in the transition state. The magnitude of this KIE can distinguish between concerted and stepwise mechanisms.
Secondary β-Deuterium KIEs: Deuterium substitution at carbon atoms adjacent to the reaction center (β-position) provides information about changes in steric congestion or hyperconjugation during the transition state. For classical SN2 reactions, these typically show normal KIEs (kₕ/k_D > 1) due to loosening of C-H bonds in the transition state as the carbon becomes more planar.
Leaving Group KIEs: Isotopic labeling of the leaving group (e.g., ³⁵S vs. ³²S) can reveal the extent of bond cleavage in the transition state. For frontside attack mechanisms, where different steric and electronic constraints might apply, these KIEs may show distinct patterns compared to classical backside attack.
Recent advances in KIE measurement, particularly through competitive experiments analyzed by NMR or MS, have enhanced precision while reducing the required quantities of isotopic materials [52]. These approaches allow researchers to measure KIEs with high accuracy even for complex biological systems or low-yield reactions.
Research Reagent Solutions for Isotope Labeling Studies
The successful implementation of isotope labeling studies requires specialized reagents and materials designed for precise isotopic incorporation and detection.
Table: Essential Research Reagents for Isotope Labeling Studies
| Reagent/Material | Isotopic Composition | Function in Mechanistic Studies | Key Applications |
|---|---|---|---|
| ¹³C-Labeled Substrates | ¹³C (99%) | Carbon tracing in reaction pathways | Tracking carbon fate in proposed SN2X mechanisms |
| ¹⁵N-Labeled Amines/Nitro Compounds | ¹⁵N (99%) | Nitrogen atom tracking | Studying amine participation in substitution reactions |
| Deuterated Solvents | ²H (99.8%) | NMR solvent for isotopic analysis | Solvent isotope effects, reaction kinetics |
| SILAC Kits | ¹³C₆/¹⁵N₂-lysine, ¹³C₆/¹⁵N₄-arginine | Metabolic protein labeling | Enzyme turnover studies in mechanistic contexts |
| TMT/Isobaric Tags | ¹³C, ¹⁵N in various configurations | Multiplexed quantitative proteomics | Simultaneous monitoring of multiple reaction pathways |
| Isotope-Coded Affinity Tags | ²H/¹³C-encoded biotin tags | Enrichment and quantification of reactive species | Trapping and identifying reaction intermediates |
| KIE Standard Kits | Position-specific ²H, ¹³C, ¹⁵N labels | Internal standards for kinetic studies | Accurate KIE measurements for transition state analysis |
These specialized reagents enable researchers to design sophisticated experiments that probe the intimate details of reaction mechanisms. For example, position-specific ¹³C-labeled substrates allow precise tracking of individual atoms through complex reaction sequences, while isotopically encoded affinity tags facilitate the isolation and identification of low-abundance intermediates that might be undetectable by conventional methods.
The selection of appropriate isotopic purity is critical, as lower isotopic enrichment can complicate data interpretation due to natural abundance contributions. For most mechanistic studies, isotopic enrichments of 98-99% are recommended to minimize these complications. Additionally, the position of isotopic incorporation must be carefully considered—whether uniform labeling throughout the molecule or specific labeling at particular atomic positions best addresses the mechanistic question under investigation.
Analytical Techniques for Isotopic Analysis
Mass Spectrometry-Based Approaches
Mass spectrometry has emerged as a cornerstone technology for isotopic analysis in mechanistic studies due to its exceptional sensitivity, specificity, and compatibility with various separation techniques [51] [50]. Several MS configurations are particularly relevant for investigating reaction mechanisms:
LC-MS/MS with Multiple Reaction Monitoring (MRM): This approach combines liquid chromatography separation with tandem mass spectrometry detection, providing exceptional specificity for target analytes [51]. In MRM mode, a precursor ion is selected in the first quadrupole, fragmented in the second quadrupole via collision-induced dissociation (CID), and specific product ions are analyzed in the third quadrupole. The combination of retention time and precursor→product ion transition provides a highly specific analytical method ideal for tracking isotopically labeled compounds in complex mixtures.
Isotope Dilution Mass Spectrometry (IDMS): This quantitative approach uses authentic stable isotope-labeled analogs as internal standards, spiked into samples at known concentrations [51] [50]. The response ratio between the analyte and labeled compound is interpolated onto a standard curve to calculate absolute amounts. IDMS provides superior accuracy and precision because the isotopic internal standard experiences nearly identical analytical behavior as the native compound, correcting for variations in sample preparation, matrix effects, and instrument response.
High-Resolution Mass Spectrometry (HRMS): Instruments such as Orbitrap and FT-ICR mass spectrometers provide sufficient mass resolution and accuracy to resolve isotopic fine structure, enabling unambiguous determination of elemental composition and isotopic incorporation. This capability is particularly valuable for identifying unexpected reaction products or intermediates that may form during mechanistic studies.
Nuclear Magnetic Resonance Spectroscopy
NMR spectroscopy complements MS-based approaches by providing structural information alongside isotopic analysis [48]. Specific NMR techniques valuable for mechanistic studies include:
¹³C NMR: Direct detection of ¹³C-labeled compounds provides information about the chemical environment of specific carbon atoms, enabling researchers to track the fate of individually labeled positions through reactions.
²H NMR: Although deuterium has lower intrinsic sensitivity than ¹H, the absence of background signals in natural abundance samples makes ²H NMR particularly useful for monitoring deuterium incorporation at specific molecular positions.
INEPT and DEPT Experiments: These polarization transfer techniques enhance the sensitivity of low-abundance nuclei like ¹³C and ¹⁵N by transferring polarization from protons, enabling detection of isotopically labeled compounds at lower concentrations.
The combination of MS and NMR approaches provides a powerful orthogonal validation strategy for isotopic labeling studies, with MS offering superior sensitivity and quantification capabilities, while NMR provides unparalleled structural information about the labeling patterns.
Isotope labeling methodologies represent indispensable tools in the mechanistic investigation of chemical reactions, particularly for elucidating complex processes such as the proposed frontside attack SN2X mechanism. Through strategic application of isotopic labels, combined with sophisticated analytical techniques including mass spectrometry and NMR spectroscopy, researchers can track atomic movements, quantify kinetic parameters, and determine stereochemical outcomes with unprecedented precision.
The continuing evolution of isotope labeling strategies, including multiplexed isobaric tagging, advanced kinetic isotope effect measurements, and computational integration of isotopic data, promises to further enhance our ability to decipher complex reaction mechanisms. As these methodologies become increasingly sophisticated and accessible, they will undoubtedly uncover new mechanistic insights and potentially reveal previously unrecognized reaction pathways in organic and biological chemistry.
For researchers investigating novel substitution mechanisms like the SN2X pathway, a multidisciplinary approach combining multiple isotope labeling strategies with theoretical calculations offers the most powerful path toward definitive mechanistic elucidation. The integration of experimental isotopic data with computational models provides a rigorous framework for testing mechanistic hypotheses and refining our understanding of fundamental chemical processes.
The recent discovery of isocyanides as potent nucleophiles in SN2 reactions with alkyl halides has introduced a transformative three-component methodology for amide bond formation. This in-depth technical guide details the reaction mechanism, optimized protocols, and broad substrate scope of this novel transformation, framing it within the context of unconventional nucleophilic substitution pathways. Supported by quantitative data and experimental workflows, this review underscores the method's significant implications for pharmaceutical synthesis, particularly in enabling late-stage functionalization and expanding accessible chemical space beyond classical amide coupling approaches.
The nucleophilic substitution (SN2) reaction represents a cornerstone of organic synthesis, enabling the predictable formation of carbon-heteroatom bonds through a concerted backside attack mechanism that proceeds with inversion of configuration [3] [1]. Traditional SN2 reactions employ nucleophiles centered on heteroatoms such as oxygen, nitrogen, sulfur, or phosphorus, while carbon-based nucleophiles have been predominantly limited to cyanide and other stabilized carbanions [55] [56].
Isocyanides, characterized by their unique electronic structure featuring a carbon-centered lone pair in a σ-orbital and a vacant π* orbital, have been extensively exploited in radical chemistry and multicomponent reactions like the Passerini and Ugi reactions [55] [57]. Despite this versatility, their potential as nucleophiles in SN2 reactions remained largely unexplored until recently. The discovery that isocyanides efficiently participate in SN2 reactions with alkyl halides, followed by in situ hydrolysis of the intermediate nitrilium ion, provides a novel three-component pathway to highly substituted secondary amides [55] [58].
This case study examines the mechanism, optimization, scope, and pharmaceutical applications of the isocyanide SN2 reaction, positioning it within broader investigations of nucleophilic substitution mechanisms. The reaction conceptually presents an Umpolung amide carbanion synthon (R-NHC(-)=O), offering a complementary approach to classical amide coupling reactions that typically require activated carboxylic acid derivatives [55] [56].
Reaction Mechanism and Stereochemical Considerations
The SN2 Mechanism: Classical Understanding
The SN2 mechanism proceeds through a single, concerted step in which nucleophilic attack and leaving group departure occur synchronously [3] [59]. This bimolecular reaction follows second-order kinetics, with rates dependent on both nucleophile and substrate concentrations [24]. The reaction proceeds exclusively through backside attack, where the nucleophile approaches the electrophilic carbon 180° opposite the leaving group, resulting in inversion of configuration at the reaction center [1].
The transition state features a trigonal bipyramidal geometry with partial bonds to both the incoming nucleophile and departing leaving group [3]. This mechanistic pathway is highly sensitive to steric effects, proceeding most rapidly with methyl and primary alkyl halides, slower with secondary substrates, and being essentially prohibitive with tertiary halides due to steric hindrance [3] [24].
Isocyanide Nucleophilicity and the SN2X Pathway
Isocyanides exhibit unique nucleophilic behavior due to their electronic structure, featuring both a high-energy σ-type highest occupied molecular orbital (HOMO) and a low-energy π-type lowest unoccupied molecular orbital (LUMO) centered on the same carbon atom [55]. This ambivalent character enables them to function as potent carbon nucleophiles in SN2 reactions, despite their limited prior exploration in this role.
The isocyanide SN2 reaction proceeds through a concerted mechanism analogous to classical SN2 pathways, but with subsequent trapping of the initial substitution product:
Diagram 1: Isocyanide SN2 reaction mechanism with hydrolysis
The initial SN2 step generates a nitrilium ion intermediate, which undergoes subsequent hydrolysis to yield the stable amide product. This three-component process (isocyanide + alkyl halide + water) effectively delivers secondary amides through an alternative disconnection strategy that bypasses traditional amine-acid coupling [55].
Experimental Optimization and Methodology
Reaction Development and Condition Optimization
Initial investigation of the isocyanide SN2 reaction employed p-chlorobenzyl isocyanide and benzyl bromide as model substrates, with reaction progress monitored via mass spectrometry and TLC analysis [55]. Through extensive high-throughput experimentation (HTE) in 96, 48, and 24-well formats, researchers systematically evaluated stoichiometry, solvent systems, temperature profiles, and catalytic additives.
Critical optimization findings revealed:
- Microwave heating at 105°C for 3 hours significantly improved yields
- A 1:2 ratio of isocyanide to alkyl halide proved optimal
- Acetonitrile as solvent provided superior solubility and reaction efficiency
- Addition of 20 mol% potassium iodide enhanced conversion by converting chloride leaving groups to more reactive iodide species
- 2 equivalents of K₂CO₃ as base and 1 equivalent of water were essential for hydrolysis [55]
Phase transfer catalysts were screened (16 varieties), with only modest improvements observed. The presence of water was found critical for the hydrolysis step but required careful stoichiometric control to prevent premature isocyanide hydrolysis [55].
Standard Experimental Protocol
Optimized General Procedure: In a microwave vial equipped with a stir bar, combine the isocyanide (1.0 mmol), alkyl halide (2.0 mmol), potassium iodide (0.2 mmol, 20 mol%), and anhydrous potassium carbonate (2.0 mmol) in anhydrous acetonitrile (5 mL). Add water (1.0 mmol, 18 μL) and seal the vial. Heat the reaction mixture under microwave irradiation at 105°C for 3 hours with stirring. Monitor reaction completion by TLC or LC-MS. Upon completion, dilute the mixture with ethyl acetate (20 mL) and wash with brine (2 × 10 mL). Dry the organic layer over anhydrous sodium sulfate, filter, and concentrate under reduced pressure. Purify the crude product by flash chromatography on silica gel to obtain the pure amide product [55].
Substrate Scope and Limitations
Electrophile Scope
The reaction demonstrates remarkable breadth in compatible alkyl halide structures, as summarized in Table 1.
Table 1: Alkyl Halide Scope in Isocyanide SN2 Reactions
| Halide Class | Example Structures | Yield Range | Key Limitations |
|---|---|---|---|
| Simple Alkyl | Methyl iodide, Ethyl bromide | 21a: 45% | β-Branched alkyl groups unreactive |
| Benzyl | Benzyl bromide, p-Cl-benzyl chloride | 1a, 3a, 4a, 6a: 40-65% | Electron-withdrawing groups well-tolerated |
| Allyl | Allyl bromide | 5a: 52% | Stabilized by conjugated transition state |
| Heteroaromatic | Benzimidazole, pyrazole, thiophene derivatives | 7a-9a, 15a: 35-60% | Various heterocycles compatible |
| Bifunctional | 4-Formylbenzyl chloride, bifunctional aromatics | 13a, 15a, 18a: 32-55% | Aldehyde functionality preserved |
Leaving group reactivity followed the classical trend I > Br > Cl, with iodides generally providing superior yields. Steric effects significantly influenced reactivity, with methyl and primary halides demonstrating excellent conversion, secondary halides showing moderate reactivity, and tertiary halides being essentially unreactive due to steric hindrance [55]. Notably, bifunctional substrates containing additional reactive handles such as aldehydes (15a, 18a) were well-tolerated, enabling further derivatization.
Isocyanide Scope
The nucleophile scope encompasses diverse isocyanide structures, as detailed in Table 2.
Table 2: Isocyanide Scope in SN2 Reactions
| Isocyanide Class | Example Structures | Yield Range | Notable Features |
|---|---|---|---|
| Aliphatic | Adamantyl, cyclohexyl | 27a, 42a: 40-60% | Adamantyl isocyanide is solid, bench-stable |
| Benzylic | p-Cl-benzyl, p-OMe-benzyl | 23a-25a, 29a-30a: 45-65% | Electronic modifications tolerated |
| Aromatic | Phenyl, naphthyl | 31a, 33a-37a: 40-70% | Sterically hindered ortho-substituents compatible |
| Heteroaromatic | Pyridyl, furyl | 26a, 28a, 32a: 35-55% | Nitrogen-containing heterocycles viable |
| Amino Acid-Derived | Valine, leucine derivatives | 40a, 41a: 40-50% | Potential for peptide mimetics |
Isocyanides with basic side chains underwent double alkylation to form quaternary ammonium salts (38a, 39a). α-Amino acid-derived isocyanides reacted efficiently, opening routes to peptide-like architectures. The solid, bench-stable nature of adamantyl isocyanide makes it particularly valuable for practical applications [55].
Pharmaceutical Applications
Late-Stage Functionalization
The method demonstrates significant utility in late-stage functionalization of complex pharmaceuticals. Using the alpha-blocker phenoxybenzamine (Dibenzyline) as a case study, reaction with adamantyl isocyanide provided the corresponding amide derivative (22a) in 40% yield, demonstrating compatibility with existing drug architectures and enabling direct diversification of pharmaceutical scaffolds [55].
Gram-Scale Synthesis and Industrial Relevance
Gram-scale reactions performed with fair yields confirm the robustness and scalability of this transformation for potential industrial applications [55]. The ability to incorporate isotope-labeled carboxy groups via isocyanide building blocks further enhances its utility in pharmaceutical development, particularly for metabolic studies or tracer synthesis.
The reaction nearly doubles the accessible chemical space compared to classical amide coupling reactions, as it employs different bond disconnections and building blocks [55] [58]. This expansion is particularly valuable for diversity-oriented synthesis in drug discovery campaigns.
Research Reagent Solutions
Table 3: Essential Research Reagents for Isocyanide SN2 Reactions
| Reagent | Function | Optimization Notes |
|---|---|---|
| Alkyl Halides | Electrophilic substrate | Iodides > Bromides > Chlorides; Methyl, primary, allylic, benzylic preferred |
| Isocyanides | Carbon nucleophile | Aliphatic, aromatic, heteroaromatic, amino acid-derived variants viable |
| Potassium Iodide | Leaving group activator | 20 mol%; converts chlorides to more reactive iodides in situ |
| Potassium Carbonate | Base | 2.0 equivalents; inorganic base essential for reaction efficiency |
| Acetonitrile | Solvent | Anhydrous conditions preferred; optimal solvent in HTE screening |
| Water | Hydrolysis agent | 1.0 equivalent; stoichiometric control critical to prevent premature hydrolysis |
Comparative Analysis with Classical Amide Synthesis
The traditional approach to amide bond formation relies on coupling between nucleophilic amines and electrophilic carboxylic acid derivatives, typically requiring pre-activation of the acid component with reagents such as thionyl chloride, oxalyl chloride, or carbodiimides [55]. These activation steps often involve aggressive reagents, generate stoichiometric waste, and present challenges in functional group compatibility.
In contrast, the isocyanide SN2 approach offers several distinct advantages:
- Alternative Bond Disconnection: Utilizes isocyanides and alkyl halides as orthogonal building blocks
- Reduced Waste: Minimizes stoichiometric activator waste
- Broad Commercial Availability: Both isocyanides and alkyl halides are readily accessible
- Functional Group Tolerance: Compatible with various protected functional groups
- Telescoped Process: Combines carbon-carbon bond formation with hydrolysis in one pot
The reaction conceptually provides an Umpolung synthon equivalent to R-NHC(-)=O, representing a polarity-reversed approach to amide construction compared to classical strategies [55] [56].
The isocyanide SN2 reaction represents a significant expansion of nucleophilic substitution chemistry, transforming isocyanides from specialized multicomponent reaction substrates into versatile nucleophiles for amide synthesis. Its broad substrate scope, operational simplicity, and compatibility with complex molecule diversification establish it as a valuable addition to the synthetic chemistry toolbox.
Within the broader context of SN2 mechanism research, this reaction exemplifies the continued potential for discovering new reactivities within seemingly well-established reaction classes. The unique three-component design, combining nucleophilic substitution with subsequent hydrolysis, demonstrates how mechanistic understanding can guide the development of synthetically useful transformations.
Future developments will likely focus on expanding stereoselective variants, developing heterogeneous or flow-based protocols for improved scalability, and further exploiting the reaction's potential in biomolecule modification and pharmaceutical synthesis. As the accessibility and diversity of isocyanide building blocks continue to improve, this methodology promises to see expanded adoption across synthetic chemistry disciplines.
Reaction Parameters and Structural Factors Influencing Substitution Pathways
The bimolecular nucleophilic substitution (SN2) reaction constitutes one of the most widely-used organic chemistry reactions, both in chemistry and biology [9]. This reaction mechanism exhibits exceptional sensitivity to the molecular structure of the substrate, particularly the degree of branching at the electrophilic carbon center. This whitepaper examines the steric effects imposed by alkyl substituents that govern the characteristic reactivity hierarchy of methyl > primary > secondary > tertiary substrates within the context of advanced SN2 reaction mechanism research.
The fundamental SN2 mechanism involves a concerted process wherein nucleophilic attack and leaving group departure occur simultaneously through a single transition state [3] [60]. This reaction pathway demands precise geometric alignment where the nucleophile must approach the electrophilic carbon from 180° relative to the carbon-leaving group (C–LG) bond, a trajectory known as backside attack [3]. The transition state adopts a trigonal bipyramidal geometry with partial bonds forming to the nucleophile and breaking to the leaving group [61]. It is this specific spatial requirement that renders the SN2 mechanism particularly vulnerable to steric congestion around the reaction center.
Mechanistic Basis for Steric Effects
The Backside Attack Imperative
The SN2 mechanism proceeds through a concerted backside attack of the nucleophile upon the alkyl halide [3]. The nucleophile approaches the electrophilic carbon from the side opposite the leaving group, donating an electron pair into the antibonding (σ*) orbital of the C–LG bond [3]. This electron donation simultaneously weakens the C–LG bond while forming the new C–Nu bond, culminating in a single transition state without intermediates [61].
The requirement for backside attack creates a sterically congested transition state featuring five groups interacting with the central carbon: the incoming nucleophile, the departing leaving group, and three substituents [62]. When these three substituents are small hydrogen atoms, minimal steric repulsion occurs with the approaching nucleophile. However, as hydrogen atoms are replaced with progressively larger alkyl groups (R groups), steric repulsion increases dramatically, impeding the nucleophile's access to the reaction center [62].
Molecular Orbital Considerations
The SN2 reaction involves nucleophile donation into the σ* orbital of the C–LG bond, which resides at 180° to the bond itself [3]. This molecular orbital orientation mandates the backside approach trajectory. As alkyl substituents increase in size and number around the electrophilic carbon, they effectively shield the σ* orbital from nucleophilic approach, reducing orbital overlap efficiency and increasing the activation energy required to reach the transition state [62].
Quantitative Reactivity Hierarchy
Experimental data consistently demonstrates that SN2 reaction rates decrease significantly as substitution increases at the electrophilic carbon. The table below summarizes the relative rates for different substrate classes:
Table 1: Relative Rates of SN2 Reactions for Different Alkyl Halides
| Substrate Type | Electrophilic Carbon Environment | Relative Rate | Steric Character |
|---|---|---|---|
| Methyl | CH₃–X | ~120 [61] | Minimal hindrance |
| Primary | R–CH₂–X | ~0.03-0.04 [61] | Moderate hindrance |
| Secondary | R₂CH–X | ~0.00002 [61] | Significant hindrance |
| Tertiary | R₃C–X | No reaction observed [62] | Complete blockage |
The dramatic rate reduction reflects the exponential relationship between steric bulk and activation energy. Branching at carbons adjacent to the electrophilic center also diminishes reaction rates, though to a lesser extent than substitution at the reaction center itself [62]. For example, 2-methyl-1-bromopropane reacts significantly slower than 1-bromopropane due to the methyl group on the neighboring carbon [62].
Experimental Characterization Methods
Kinetic Analysis Protocols
Rate Law Determination establishes the molecularity of the substitution process. For authentic SN2 pathways, the reaction displays second-order kinetics, with rate dependent on both nucleophile and substrate concentrations [3] [60]. The experimental protocol involves:
- Preparation of separate nucleophile and substrate solutions at varying concentrations (typically 0.01-0.1 M range)
- Reaction initiation by mixing thermally equilibrated solutions
- Reaction monitoring via timed aliquots quenched in appropriate solvent
- Product quantification using GC, HPLC, or spectroscopic methods
- Rate constant calculation from plots of concentration versus time
The confirmed rate law takes the form: Rate = k [alkyl halide] [nucleophile], where k is the second-order rate constant [61].
Stereochemical Analysis
Inversion of configuration serves as a hallmark diagnostic for the SN2 mechanism [3]. The experimental approach employs:
- Preparation of chiral substrates starting from enantiomerically pure precursors
- Reaction with nucleophiles under SN2-favoring conditions (polar aprotic solvents, good nucleophiles)
- Product stereochemistry analysis using polarimetry, chiral HPLC, or NMR with chiral shift reagents
The characteristic Walden inversion occurs because the nucleophile's backside approach inverts the stereocenter's configuration during the transition state [3] [63]. This stands in contrast to SN1 reactions, which proceed through planar carbocation intermediates and produce racemic mixtures [61] [63].
Table 2: Experimental Protocols for SN2 Mechanism Characterization
| Method | Key Procedure | SN2 Diagnostic Outcome | Research Considerations |
|---|---|---|---|
| Kinetic Analysis | Variation of nucleophile and substrate concentrations | Second-order rate law: Rate = k[alkyl halide][nucleophile] [61] | Requires precise concentration control and reaction quenching |
| Stereochemical Analysis | Use of chiral substrates with defined configuration | Complete inversion of stereochemistry at reaction center [3] [63] | Requires enantiomerically pure starting materials and chiral analysis methods |
| Competitive Kinetics | Comparison of relative rates across substrate classes | Reactivity order: Methyl > Primary > Secondary >> Tertiary [61] [62] | Normalization for electronic effects is critical |
| Solvent Effects | Reaction in polar aprotic versus protic solvents | Rate acceleration in polar aprotic solvents (e.g., DMSO, DMF) [63] | Controls for ion pairing and nucleophile solvation |
Computational Analysis
Modern computational studies provide atomic-level insight into steric effects on SN2 reactions. The activation strain model analyzes activation barriers as the sum of energies required to distort reactants into transition state geometries plus the interaction energies between the deformed reactants [9]. Key computational protocols include:
- Geometry optimization of reactants, transition states, and products using density functional theory (DFT)
- Transition state validation through frequency calculations (single imaginary frequency)
- Intrinsic reaction coordinate (IRC) calculations to connect transition states to corresponding minima
- Activation strain analysis to decompose energy barriers into strain and interaction components
These methods reveal how increasing alkyl substitution elevates transition state strain energy, rationalizing the observed reactivity hierarchy [9].
Research Reagent Solutions
Table 3: Essential Research Reagents for SN2 Steric Studies
| Reagent Category | Specific Examples | Research Function | Steric Relevance |
|---|---|---|---|
| Model Nucleophiles | Halide ions (I⁻, Br⁻, Cl⁻), CN⁻, N₃⁻, CH₃O⁻ | Standardized nucleophilic strength assessment | Steric demands vary with nucleophile size |
| Sterically-Diverse Substrates | CH₃-I, CH₃CH₂-Br, (CH₃)₂CH-Br, (CH₃)₃C-Br | Systematic evaluation of steric hierarchy | Progressive crowding at reaction center |
| Polar Aprotic Solvents | Dimethyl sulfoxide (DMSO), Dimethylformamide (DMF), Acetonitrile | Nucleophile activation by reducing solvation | Minimizes solvent coordination to nucleophile |
| Chiral Probes | (S)-2-bromobutane, enantiomerically pure substrates | Stereochemical trajectory analysis | Confirms inversion mechanism characteristic of SN2 |
Implications for Drug Development
The steric hierarchy of SN2 reactivity has profound implications for pharmaceutical design and prodrug strategies. Medicinal chemists leverage this understanding to:
- Design hydrolytically stable compounds by incorporating steric shielding around susceptible electrophilic centers
- Create prodrug activation systems where enzymatic removal of steric blocking groups enables intramolecular SN2 cyclization or reaction
- Predict metabolic pathways involving biomolecular substitution reactions
- Optimize synthetic routes for active pharmaceutical ingredients (APIs) where SN2 steps are employed
Understanding steric constraints on nucleophilic substitution enables researchers to modulate compound reactivity, stability, and activation profiles for enhanced therapeutic efficacy.
The reactivity hierarchy of methyl > primary > secondary > tertiary for SN2 reactions represents a fundamental principle in organic chemistry with far-reaching implications. This steric gradient originates from the concerted backside attack mechanism that necessitates nucleophilic approach to a increasingly shielded electrophilic center. Quantitative kinetic studies reveal exponential rate diminution with increasing substitution, while stereochemical analysis confirms the inversion pathway characteristic of this bimolecular process. This understanding provides pharmaceutical researchers with strategic approaches for molecular design targeting enhanced stability, controlled activation, and predictable metabolic fate. Further research continues to explore the frontier of steric effects, including computationally-guided predictions and applications in sophisticated drug delivery systems.
Understanding nucleophile characteristics—strength, solvation, and steric demand—is fundamental to predicting and controlling organic reactivity. This knowledge forms the bedrock of synthetic design in fields ranging from materials science to pharmaceutical development. For decades, the reaction landscape has been dominated by the classical bimolecular nucleophilic substitution (SN2) mechanism, characterized by its concerted nature and definitive stereochemical inversion resulting from a backside attack [3]. However, recent research has revealed a more complex picture with the discovery of the halogenophilic nucleophilic substitution (SN2X) pathway, a distinct mechanism that proceeds through a frontside attack and exhibits contrasting sensitivity to steric and solvation effects [4] [64]. This whitepaper provides an in-depth technical guide to nucleophile characteristics, framing classical principles within the context of this emerging reaction paradigm. A comparative analysis of these mechanisms is crucial for researchers aiming to harness the unique selectivity and efficiency of the SN2X pathway, particularly for challenging transformations involving sterically hindered substrates in drug development.
Foundational Principles of Nucleophile Strength
Nucleophilicity is defined as the ability of a species to donate an electron pair to an electrophile, a process kinetically measured by comparing reaction rates [65]. A stronger nucleophile leads to a faster reaction rate. Several key factors independently govern nucleophile strength, and their interplay ultimately determines reactivity.
Charge and Electron Density
A nucleophile's strength is directly proportional to its electron density. Species with a full negative charge are significantly better nucleophiles than their neutral counterparts. This is because the increased electron density enhances the species' willingness to donate its electrons. For example, the hydroxide ion (HO⁻) is a much stronger nucleophile than water (H2O), and an amide ion (NH2⁻) is stronger than ammonia (NH3) [66]. A fundamental rule is that the conjugate base of a species is always a better nucleophile [65].
Electronegativity and Polarizability
The role of the nucleophilic atom's identity is critical and operates through two primary trends:
- Across a Row: Moving from left to right across the periodic table, electronegativity increases. Atoms with higher electronegativity hold their electrons more tightly, making them less available for donation. Consequently, nucleophilicity decreases with increasing electronegativity across a period. The trend is: C⁻ > N⁻ > O⁻ > F⁻ [67].
- Down a Group: Moving down a group in the periodic table, atomic size and polarizability increase. Larger atoms have electron clouds that are more easily distorted (polarized), which facilitates bond formation with the electrophilic carbon. Therefore, nucleophilicity increases down a group, particularly in polar protic solvents. The trend for halogens is: I⁻ > Br⁻ > Cl⁻ > F⁻ [65] [67].
Solvation Effects
The solvent medium can profoundly influence nucleophile strength, sometimes even reversing the trends predicted by basicity alone.
- Polar Protic Solvents (e.g., H2O, ROH) contain hydrogens bonded to oxygen or nitrogen. These solvents can form strong hydrogen bonds with anionic nucleophiles, stabilizing them and reducing their reactivity [65] [66]. Smaller anions (e.g., F⁻) are more strongly solvated (and thus more deactivated) than larger anions (e.g., I⁻), which are less effectively shielded by the solvent. This explains the high nucleophilicity of iodide in alcohols [65].
- Polar Aprotic Solvents (e.g., DMSO, acetone) lack acidic hydrogens and cannot hydrogen bond effectively with nucleophiles. In these solvents, anions are "naked" and more reactive. Here, nucleophilicity correlates well with basicity, making fluoride ion the strongest nucleophile among the halides [65].
Table 1: Key Factors Determining Nucleophile Strength
| Factor | Trend | Rationale | Example |
|---|---|---|---|
| Charge | Negatively charged > Neutral | Higher electron density increases electron-donating ability. | HO⁻ > H2O |
| Electronegativity (Across a Row) | Nucleophilicity decreases with increasing electronegativity | More electronegative atoms hold electrons more tightly. | N⁻ > O⁻ > F⁻ |
| Polarizability (Down a Group) | Nucleophilicity increases down a group (in protic solvents) | Larger electron clouds are more easily distorted to form new bonds. | I⁻ > Br⁻ > Cl⁻ > F⁻ |
| Solvent (Protic vs. Aprotic) | Protic solvents solvate and shield small anions; Aprotic solvents do not. | Solvation shell in protic solvents hinders nucleophile approach. | In Protic: I⁻ > F⁻ In Aprotic: F⁻ > I⁻ |
Table 2: Qualitative Nucleophile Strength Ranking in Polar Protic Solvents [65] [67]
| Strength Category | Representative Nucleophiles |
|---|---|
| Very Good | I⁻, HS⁻, RS⁻ |
| Good | Br⁻, HO⁻, RO⁻, NC⁻, N3⁻ |
| Fair | NH3, Cl⁻, F⁻, RCO2⁻ |
| Weak | H2O, ROH |
Steric Demand and Its Mechanistic Implications
Steric hindrance refers to the physical blockade of a reactive center by bulky substituents, which can dramatically slow down reaction rates. The demand for a "sterically unhindered" path is one of the most defining differentiators between classical and emerging substitution mechanisms.
Steric Hindrance in the SN2 Mechanism
The classical SN2 mechanism proceeds via a concerted backside attack, where the nucleophile must directly approach the electrophilic carbon from the side opposite the leaving group [3]. This process results in a pentacoordinated transition state where the carbon is simultaneously partially bonded to the nucleophile, the leaving group, and its three substituents [68]. The crowding in this transition state is severe. As a result, SN2 reaction rates are extremely sensitive to steric effects and follow a well-established reactivity order: Methyl > Primary > Secondary >> Tertiary [68] [3]. Tertiary alkyl halides are essentially unreactive in SN2 reactions due to this steric blockade [3]. Bulky nucleophiles (e.g., tert-butoxide) are also less reactive than less hindered ones (e.g., methoxide) for the same reason [65].
The SN2X Mechanism and Reduced Steric Demand
The recently characterized SN2X (halogenophilic nucleophilic substitution) reaction represents a paradigm shift. It follows a frontside attack pathway, where the nucleophile initially interacts with the halogen leaving group (or chalcogen in the related SN2Ch reaction), forming a pro-chiral anion intermediate [4] [64]. This distinct pathway, with its different transition state geometry, is not prone to the same steric hindrance that governs SN2 reactions [64]. This key difference allows the SN2X pathway to proceed efficiently with sterically hindered tertiary halides, substrates that are completely unreactive under classical SN2 conditions [64]. The parameter of relative halogenophilicity (H) has been developed to quantitatively characterize the intrinsic nature of these reactions [4].
Diagram 1: Steric Demand in SN2 vs. SN2X
Quantitative Data and Experimental Methodology
A quantitative understanding is essential for researchers to apply these concepts in practical settings, from predicting reaction outcomes to designing novel synthetic protocols.
Quantitative Nucleophilicity Scales
Nucleophilicity is measured kinetically. A standard scale is based on the relative rates of the SN2 reaction with methyl iodide in methanol at 25°C [65]. This provides a numerical basis for comparing nucleophile strength.
Table 3: Relative Nucleophilicity in Methanol (Reference: CH3OH = 1.0) [65]
| Nucleophile | Relative Reactivity | Nucleophile | Relative Reactivity |
|---|---|---|---|
| CN⁻ | 1260 | Br⁻ | 175 |
| I⁻ | 400 | Cl⁻ | 6.7 |
| HO⁻ | 100 | CH3OH | 1.0 |
Quantifying SN2 versus SN2X Competition
The discovery of the SN2X pathway necessitates methods to distinguish it from SN2. Kuo et al. (2024) developed a quantitative procedure using kinetic simulations to measure the halogenophilic percentage (X%)—the fraction of the reaction proceeding via the SN2X pathway [4]. They also introduced the parameter relative halogenophilicity (H), which quantifies the intrinsic characteristics of the SN2X reaction and correlates with established physical organic chemistry principles like the Hammett and Mayr postulates [4].
Experimental Protocol for Differentiating SN2 and SN2X Pathways
The following methodology, adapted from seminal research, allows for the experimental investigation and quantification of these competing pathways [4] [64].
Objective: To determine the percentage of halogenophilic substitution (X%) in the nucleophilic substitution of a sterically hindered, enantiomerically enriched tertiary alkyl halide.
Principle: The SN2 pathway is stereospecific, leading to inversion of configuration. In contrast, the SN2X pathway proceeds via a pro-chiral anion intermediate, which can lead to racemization or a different stereochemical outcome. Analyzing the stereochemistry of the product allows for the quantification of each pathway's contribution.
Materials:
- Substrate: Enantiomerically pure tertiary alkyl halide (e.g., (R)- or (S)-enantiomer).
- Nucleophile: A strong, well-characterized nucleophile (e.g., a thiolate or a secondary amine).
- Solvent: An appropriate polar aprotic solvent (e.g., DMF or acetone) to ensure nucleophile reactivity.
- Analysis Equipment: Chiral HPLC or GC for precise measurement of product enantiomeric excess (e.e.).
Procedure:
- Reaction Setup: Dissolve the enantiomerically pure tertiary alkyl halide (e.g., 0.1 mmol) and the nucleophile (e.g., 0.12 mmol) in the chosen solvent under an inert atmosphere at a controlled temperature (e.g., 25°C).
- Reaction Monitoring: Monitor the reaction progress by TLC or LC/MS until completion.
- Product Isolation: Upon completion, quench the reaction and isolate the substitution product using standard work-up and purification techniques (e.g., extraction, chromatography).
- Stereochemical Analysis: Determine the enantiomeric excess (e.e.) of the purified product using chiral HPLC or GC.
- Data Analysis & Kinetic Simulation:
- Calculate the observed stereochemical outcome (e.g., % inversion, % retention, % racemization).
- Input the experimental data into a kinetic simulation model that accounts for the rates of the SN2 pathway (stereospecific inversion) and the SN2X pathway (formation of a racemic or scrambled product via the anion intermediate).
- The simulation fits the data to solve for the halogenophilic percentage (X%), representing the fraction of the total reaction that proceeded via the SN2X mechanism.
Diagram 2: SN2X Experimental Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Advancing research in nucleophilic substitution, particularly concerning the SN2X mechanism, requires a specific set of chemical tools and analytical resources.
Table 4: Key Research Reagent Solutions for SN2X Investigation
| Reagent / Material | Function & Rationale |
|---|---|
| Sterically Hindered Tertiary Alkyl Halides | The quintessential substrate for probing SN2X reactivity. Their inaccessibility to classical SN2 backside attack allows the frontside SN2X pathway to be isolated and studied [64]. |
| Polar Aprotic Solvents (DMSO, DMF, Acetone) | These solvents enhance nucleophile reactivity by poorly solvating anions, making them essential for achieving measurable reaction rates with sterically hindered electrophiles [65]. |
| Strong, Soft Nucleophiles (e.g., Thiolates, Azide) | Often used in discovery studies; their high polarizability can favor interaction with the halogen atom in the initial step of the SN2X mechanism [4] [67]. |
| Chiral Stationary Phase HPLC/GC Columns | Critical for determining the enantiomeric purity of starting materials and products. The stereochemical outcome is the primary data for distinguishing SN2 from SN2X pathways [4]. |
| Deuterated Solvents for NMR Analysis | Used for reaction monitoring and characterization of intermediates and regioisomers, providing structural evidence for the formation of unique species in the SN2X pathway. |
| Parameter H (Relative Halogenophilicity) | A quantitative descriptor, analogous to established nucleophilicity scales, used to predict and rationalize the propensity of a given system to undergo the SN2X reaction [4]. |
The characteristics of nucleophiles—their intrinsic strength, complex interactions with solvents, and the steric demands of their approach—are not merely academic concepts but powerful levers for controlling chemical reactivity. The classical model of the SN2 reaction, with its stringent requirements for a low-steric-demand backside attack, has been successfully augmented by the discovery of the SN2X mechanism. This frontside attack pathway, with its reduced steric demand and unique halogen-focused mechanism, overturns long-held assumptions and expands the synthetic chemist's toolbox [64]. For researchers and drug development professionals, this new understanding is pivotal. It enables the rational design of reactions involving sterically congested molecules, a common challenge in synthetic organic and medicinal chemistry. The quantitative tools now available, such as the measurement of X% and the parameter H, provide a framework for exploring this chemical space systematically [4]. Future research will undoubtedly uncover the full scope and limitations of the SN2X reaction, refine its predictive models, and explore its application in catalytic cycles and complex molecular synthesis. Re-examining other "understood" organic reactions in light of this discovery may lead to further paradigm shifts, driving innovation across the chemical sciences.
Within nucleophilic substitution reactions, the solvent is not a passive spectator but a critical determinant of reaction pathway, rate, and selectivity. For researchers investigating non-canonical pathways such as the frontside attack SN2X mechanism, a precise understanding of solvent effects is paramount. This guide provides an in-depth analysis of how polar protic and aprotic media influence the energetic landscape of nucleophilic substitutions, equipping scientists with the knowledge to design experiments and interpret results within a sophisticated mechanistic framework.
The classic SN2 mechanism is a concerted process characterized by backside nucleophilic attack, leading to inversion of configuration, while the SN1 mechanism proceeds through a stepwise pathway involving a carbocation intermediate [69] [70]. The continuum between these mechanisms, particularly for secondary substrates, often exhibits borderline characteristics where the solvent actively participates in the transition state [71]. This technical overview details the governing principles, supported by quantitative data and experimental protocols, to inform research in synthetic and medicinal chemistry.
Solvent Classification and Properties
Solvents are systematically classified based on their polarity and ability to donate hydrogen bonds, which directly dictates their interactions with solutes.
Defining Key Characteristics
- Polarity: A solvent's overall dipole moment and ability to stabilize charges, often quantified by its dielectric constant (ε). Higher dielectric constants indicate greater polarity [72].
- Hydrogen Bonding: The critical distinction lies in whether a solvent can act as a hydrogen bond donor. Polar protic solvents contain O-H or N-H bonds and can form strong hydrogen bonds with anions and lone pairs. In contrast, polar aprotic solvents lack such acidic hydrogens and cannot form strong hydrogen bonds with nucleophiles [72] [73].
Table 1: Classification and Properties of Common Laboratory Solvents
| Solvent Type | Example Solvents | Dielectric Constant (ε)* | Key Structural Feature | Hydrogen Bond Donor Capability |
|---|---|---|---|---|
| Polar Protic | Water, Methanol, Ethanol, Acetic Acid | ~78 (H₂O), ~33 (MeOH) | O-H or N-H bonds | Yes |
| Polar Aprotic | Dimethylformamide (DMF), Dimethyl Sulfoxide (DMSO), Acetonitrile (CH₃CN), Acetone | ~37 (DMF), ~47 (DMSO), ~37 (MeCN), ~21 (Acetone) | Polar bonds (e.g., C=O, S=O, C≡N) but no O-H/N-H | No |
| Nonpolar | Hexane, Benzene, Toluene | ~2 (Hexane) | C-C and C-H bonds only | No |
Note: Dielectric constant values are approximate and at ~20-25°C [72] [73].
Solvent Effects on Nucleophilic Substitution Mechanisms
Impact on the SN2 Mechanism
The SN2 reaction, being concerted, is highly sensitive to the strength of the nucleophile, which is profoundly modulated by the solvent.
- Polar Aprotic Solvents Enhance SN2 Rates: These solvents solvate cations effectively but poorly solvate anions due to the absence of hydrogen bonding. This leaves anionic nucleophiles relatively "naked" and highly reactive [74] [75] [73]. The high polarity of these solvents also helps stabilize the charged, pentacoordinate transition state without deactivating the nucleophile [74].
- Polar Protic Solvents Suppress SN2 Rates: These solvents form a strong solvation shell around anionic nucleophiles via hydrogen bonding. This "caging" effect stabilizes the nucleophile in its ground state, thereby increasing the activation energy required for the reaction and dramatically slowing the rate [75] [73]. For example, the reaction of cyanide ion with an alkyl halide can be 5000 times faster in acetonitrile (polar aprotic) than in methanol (polar protic) [73].
Impact on the SN1 Mechanism
The SN1 mechanism, involving charge separation in its rate-determining step, benefits from a different solvent environment.
- Polar Protic Solvents Strongly Favor SN1: The rate-determining step is the ionization of the substrate to form a carbocation and the leaving group. Polar protic solvents stabilize both of these charged species in the transition state through powerful ion-dipole interactions and hydrogen bonding to the leaving group [70] [73]. This stabilization significantly lowers the activation energy for the ionization step.
- Role in Subsequent Step: The solvent (e.g., H₂O, ROH) often also acts as the nucleophile in the second step of the SN1 mechanism, a process termed solvolysis [70] [73].
The Borderline Mechanistic Regime
For secondary alkyl substrates, the distinction between SN1 and SN2 is often blurred, leading to a borderline mechanism [71]. Quantum-chemical studies on systems like isopropyl chloride hydrolysis reveal a loose SN2-like mechanism with nucleophilic solvent assistance [71]. In this pathway, the solvent shell does not merely stabilize pre-formed ions but actively participates in a dissociative transition state that has significant SN1 character. This merged mechanism underscores the limitation of the binary classification and highlights the role of explicit solvent molecules in modulating the reaction coordinate.
Table 2: Comparative Summary of Solvent Effects on Nucleophilic Substitution Mechanisms
| Factor | SN1 Mechanism | SN2 Mechanism | Borderline Mechanism |
|---|---|---|---|
| Preferred Solvent | Polar Protic (e.g., H₂O, MeOH) | Polar Aprotic (e.g., DMF, DMSO) | Highly dependent on explicit solvation structure [71] |
| Effect of Solvent Polarity | Greatly accelerated by high polarity (stabilizes ions) | Moderately accelerated by polarity (stabilizes TS) | Complex, depends on solute-solvent configurational sampling [71] |
| Role of Solvent | Stabilizes ions via ion-dipole forces and H-bonding; often acts as nucleophile (solvolysis) | Poorly solvates anions, increasing nucleophilicity; stabilizes TS | Active participation in a loose, associative transition state with dissociative character [71] |
| Experimental Implication | Use protic solvents with weak nucleophiles for tertiary/substrate solvolysis | Use aprotic solvents with strong anionic nucleophiles for primary/substrate substitution | Requires advanced computational models (explicit/implicit solvation) for accurate prediction [71] |
Essential Research Reagents and Experimental Methodologies
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Studying Solvent Effects in Nucleophilic Substitution
| Reagent/Solution | Technical Function in Research |
|---|---|
| Dimethylformamide (DMF) | High-boiling polar aprotic solvent used to maximize nucleophile reactivity in SN2 screens and kinetic studies. |
| Dimethyl Sulfoxide (DMSO) | Powerful polar aprotic solvent with high dielectric constant; dissolves a wide range of organic and ionic compounds for mechanistic probing. |
| Deuterated Solvents (CD₃OD, D₂O, (CD₃)₂SO) | Protic and aprotic solvents isotopically labeled for reaction monitoring by NMR spectroscopy (e.g., kinetic isotope effects, reaction progress). |
| Isopropyl Chloride (iPrCl) | Model secondary substrate for investigating borderline SN1-SN2 mechanisms in hydrolysis/solvolysis experiments [71]. |
| Tetraalkylammonium Salts (e.g., [ⁿBu₄N]F) | Source of "naked" anions in polar aprotic media; minimizes ion-pairing effects that can confound nucleophilicity measurements. |
Computational Protocol for Borderline Mechanism Analysis
Advanced computational methods are indispensable for a molecular-level understanding of solvent effects in borderline mechanisms. The following protocol, adapted from studies on isopropyl chloride hydrolysis, provides a robust framework [71].
Step 1: System Preparation and Initial Configurations
- Substrate Selection: Choose the secondary alkyl substrate (e.g., isopropyl chloride).
- Solvent Shell Generation: Generate initial configurations of the substrate surrounded by explicit solvent molecules (e.g., H₂O). Two primary approaches are recommended:
- Top-Down (Stochastic): Use Monte Carlo (MC) simulations of the bulk condensed phase to generate statistically relevant solvent configurations around the solute [71].
- Bottom-Up (Microsolvation): Manually add explicit solvent molecules to key regions of the substrate (e.g., around the leaving group and potential nucleophilic attack sites) based on chemical intuition and molecular electrostatic potential analysis [71].
Step 2: Quantum-Chemical Calculations
- Electronic Structure Method: Employ Density Functional Theory (DFT). The M06-2X functional is recommended for its good performance in modeling reaction mechanisms and dispersion interactions [71].
- Basis Set: Use a double-zeta basis set with diffuse functions, such as aug-cc-pVDZ, for a balance of accuracy and computational cost [71] [76].
- Geometry Optimization: Locate reactants, pre-reactive complexes, transition states (TS), and products on the potential energy surface. The nature of each stationary point must be confirmed by frequency calculations (one imaginary frequency for TS, all real for minima).
- Reaction Pathway Verification: Perform Intrinsic Reaction Coordinate (IRC) calculations to confirm that the transition state correctly connects to the intended reactants and products [71].
Step 3: Solvation Modeling
- Hybrid Implicit/Explicit Model: Embed the substrate-explicit solvent cluster within a continuum implicit solvation model (e.g., PCM, SMD) to account for the bulk solvent effects beyond the first solvation shell [71].
- Convergence Testing: Systematically increase the number of explicit solvent molecules (n) until the reaction barrier (ΔH‡) converges. For water, clusters of ~9-12 molecules are often sufficient to model the key interactions [71].
Step 4: Data Analysis
- Activation Strain Analysis: Decompose the energy barrier into contributions from the substrate strain and solute-solvent interaction energy [71].
- Charge Analysis: Perform population analysis (e.g., CHELPG) to track charge distribution and leaving group stabilization throughout the reaction coordinate [71].
- Mechanistic Interpretation: Construct a More O'Ferrall–Jencks diagram to visualize the position of the transition state along the SN1-SN2 mechanistic continuum [71].
Visualizing Solvent Interactions and Mechanistic Pathways
The following diagrams, generated with DOT language, illustrate the key concepts of solvation and the resultant mechanistic outcomes.
The strategic selection of solvent media is a powerful tool for controlling reaction mechanisms in organic synthesis and biochemical analysis. Polar protic solvents favor unimolecular pathways (SN1, E1) through stabilization of ionic intermediates, while polar aprotic solvents accelerate bimolecular pathways (SN2) by enhancing nucleophile strength. For the complex frontier of frontside attack mechanisms and borderline reactions, sophisticated computational models that explicitly account for solvent molecules are essential for accurate mechanistic prediction. Mastery of these principles enables researchers to rationally design reaction conditions, optimize synthetic routes, and advance the fundamental understanding of chemical reactivity.
Leaving Group Ability and Its Relationship to Reaction Pathway
Leaving group ability is a fundamental property in organic chemistry that profoundly influences the feasibility, mechanism, and rate of nucleophilic substitution reactions. Within the context of bimolecular nucleophilic substitution (SN2) reactions, the nature of the leaving group traditionally dictates the reaction trajectory, classically proceeding via a backside attack that results in inversion of configuration at the electrophilic carbon center [3] [54]. However, emerging research on the frontside attack nucleophilic substitution (SN2X) mechanism reveals a more complex relationship, where leaving group ability can alter the very pathway of the reaction, enabling unconventional approaches to stereoselective synthesis with significant implications for pharmaceutical development [4] [36]. This technical guide examines the intrinsic and contextual factors governing leaving group ability, its quantitative assessment, and its critical role in determining reaction pathway selection between conventional SN2 and halogenophilic SN2X mechanisms.
Fundamental Principles of Leaving Group Ability
Defining Leaving Group Competence
A leaving group is a molecular fragment that departs with an electron pair during heterolytic bond cleavage in substitution and elimination reactions [77]. The general principle governing leaving group efficacy is straightforward: the weaker the base, the better the leaving group [78] [79]. This correlation exists because the leaving group must stabilize the additional electron density it acquires upon bond cleavage. Strong bases, which possess high electron density and are highly stabilized in their protonated forms, poorly stabilize additional negative charge and thus resist departure [79].
The ability of a leaving group to accommodate electron density is influenced by three key factors:
- Electronegativity: As electronegativity increases across the periodic table, basicity decreases, potentially enhancing leaving group ability [78]
- Atomic size: As atomic size increases down a periodic table group, electron density becomes more diffuse, decreasing basicity and improving leaving group ability [78]
- Resonance stabilization: Leaving groups that form resonance-stabilized structures upon departure exhibit significantly enhanced leaving ability [78]
Quantitative Assessment Using pKa
The leaving group ability can be quantitatively predicted using pKa values of their conjugate acids. The correlation follows the principle that stronger acids (lower pKa) have weaker conjugate bases, which correspond to better leaving groups [79] [77].
Table 1: Leaving Group Ability Correlated with Conjugate Acid pKa
| Leaving Group | Conjugate Acid | pKa of Conjugate Acid | Relative Ability |
|---|---|---|---|
| R-N₂⁺ | HN₂⁺ | ~4.6 | Excellent |
| R-OSO₂CF₃ (Triflate) | HOSO₂CF₃ | ~ -6 [77] | Excellent |
| R-I | HI | -10 | Very Good |
| R-OTs (Tosylate) | HOTs | ~ -2 [77] | Very Good |
| R-Br | HBr | -9 | Good |
| R-OH₂⁺ | H₃O⁺ | -1.7 | Good (when protonated) |
| R-Cl | HCl | -7 | Moderate |
| R-OCOR | RCOOH | ~5 | Poor |
| R-F | HF | 3.2 | Very Poor |
| R-OH | H₂O | 15.7 | Very Poor |
| R-OR | ROH | ~16 | Very Poor |
| R-NR₂ | R₃NH⁺ | ~10-11 | Extremely Poor |
A notable exception to the pKa correlation is fluoride (F⁻), which despite having a relatively low conjugate acid pKa of 3.2, is an extremely poor leaving group due to the exceptional strength of the C-F bond (approximately 130 kcal/mol) [79]. This demonstrates that while basicity is a primary factor, bond strength also significantly influences leaving group competence.
Leaving Groups in Conventional S_N2 Reactions
Mechanism and Stereochemical Requirements
The classic S_N2 mechanism proceeds through a concerted process in which bond formation between the nucleophile and electrophilic carbon occurs simultaneously with bond cleavage to the leaving group [3] [54]. This mechanism exhibits second-order kinetics, with the rate dependent on both substrate and nucleophile concentrations [3] [80]. The reaction proceeds through a single transition state characterized by a trigonal bipyramidal geometry, with the nucleophile and leaving group occupying apical positions [3].
A defining feature of the traditional S_N2 mechanism is the backside attack, where the nucleophile approaches the carbon center 180° from the leaving group [3] [54]. This trajectory results in inversion of configuration at chiral centers, a stereochemical outcome that serves as a diagnostic marker for this mechanism [3] [80].
Leaving Group Effects on S_N2 Reactivity
In conventional S_N2 reactions, the nature of the leaving group significantly impacts reaction rates. The relative reactivity of halide leaving groups follows the order: I⁻ > Br⁻ > Cl⁻ >> F⁻, correlating with decreasing C-X bond strength and increasing polarizability [78] [81]. Sulfonate esters, particularly triflate (OSO₂CF₃), tosylate (OTs), and mesylate (OMs), are exceptionally good leaving groups due to resonance stabilization of the anionic conjugate bases [78] [77].
Table 2: Relative Rates for S_N2 Reactions with Different Leaving Groups
| Leaving Group (X) | Relative Rate (k_rel) | Nucleophile | Conditions |
|---|---|---|---|
| Cl | 0.0074 | EtO⁻ | Not specified [77] |
| Br | 1.0 | EtO⁻ | Not specified [77] |
| I | 3.5 | EtO⁻ | Not specified [77] |
| OTs | 0.44 | EtO⁻ | Not specified [77] |
| Cl | 0.0024 | p-Thiocresolate | 40°C [77] |
| Br | 1.0 | p-Thiocresolate | 40°C [77] |
| I | 1.9 | p-Thiocresolate | 40°C [77] |
| OTs | 3.6 | p-Thiocresolate | 40°C [77] |
The data demonstrates that leaving group ability is context-dependent, varying with the nature of both the nucleophile and reaction conditions [77].
The S_N2X Mechanism: A Frontside Attack Pathway
Discovery and Defining Characteristics
Recent research has uncovered an alternative nucleophilic substitution pathway known as the halogenophilic nucleophilic substitution (SN2X) reaction [4] [36]. This mechanism exhibits a distinctly different reaction pathway compared to conventional SN2 reactions, despite often yielding identical products [4].
The SN2X mechanism proceeds through a frontside attack where the nucleophile approaches the substrate from the same side as the leaving group [36]. This stands in direct contrast to the backside attack of traditional SN2 reactions. Experimental and computational studies suggest that this unconventional trajectory is facilitated by halogen bonding interactions between the nucleophile and the leaving group [36].
Stereochemical Implications
The SN2X mechanism exhibits different stereochemical behavior compared to conventional SN2 reactions. While traditional SN2 reactions proceed with complete inversion of configuration due to backside attack, the SN2X pathway can lead to enantioconvergent outcomes, where racemic starting materials are converted to enantiomerically enriched products [36]. This stereochemical profile arises from the presence of a pro-chiral anion intermediate in the SN2X pathway, distinguishing it from the stereospecific nature of conventional SN2 reactions [4].
The enantioconvergent nature of S_N2X reactions makes this mechanism particularly valuable for asymmetric synthesis, with significant potential applications in pharmaceutical development where specific stereoisomers often exhibit different biological activities [36].
Experimental Methodologies for Studying Leaving Group Effects
Kinetic Analysis of Leaving Group Ability
Protocol 1: Determining Relative Leaving Group Competence
Reaction System Selection: Choose a standardized substrate structure (typically primary alkyl derivatives) and reaction conditions (solvent, temperature) to compare different leaving groups [77].
Nucleophile Standardization: Employ a nucleophile of sufficient strength and consistency across experiments, such as ethoxide (EtO⁻) or thiocresolate [77].
Rate Constant Determination: Monitor reaction progress using appropriate analytical techniques (NMR, GC, HPLC) to determine rate constants (k) for each leaving group [54].
Relative Rate Calculation: Normalize rates against a reference leaving group (typically bromide, k_rel = 1.0) to establish a quantitative hierarchy of leaving group ability [77].
Contextual Validation: Repeat measurements with different nucleophiles to assess the context-dependence of leaving group ability [77].
Protocol 2: Quantitative Study of Competing SN2 and SN2X Pathways
Stereochemical Probes: Utilize enantiomerically enriched substrates to distinguish between stereospecific (SN2) and enantioconvergent (SN2X) pathways [4].
Kinetic Simulations: Develop kinetic models that account for both pathways and fit experimental data to determine the halogenophilic percentage (X%) [4].
Parameter Determination: Calculate the relative halogenophilicity (H) to quantify intrinsic characteristics of S_N2X reactions [4].
Time-Dependency Analysis: Monitor X% under varying reaction conditions to identify secondary processes such as bromide-catalyzed dynamic kinetic resolution [4].
Mechanistic Probes
Stereochemical Analysis: Starting with enantiomerically pure substrates allows researchers to distinguish between inversion (SN2) and racemization or enantioconvergence (SN2X) pathways [4] [80].
Solvent Effects: Systematic variation of solvent polarity can reveal mechanistic differences, as SN2 reactions typically slow with increasing polarity for anionic nucleophiles, while SN2X behavior may show different sensitivity [9].
Computational Studies: High-level quantum mechanical calculations can map potential energy surfaces and identify transition states, providing theoretical validation for proposed mechanisms [4] [9].
Reaction Pathway Determination: SN2 versus SN2X
The interplay between leaving group ability, nucleophile characteristics, and substrate structure determines the dominant reaction pathway. The following diagram illustrates the key factors influencing mechanism selection between SN2 and SN2X pathways:
Mechanism Selection Between S_N2 and S_N2X Pathways
The competition between SN2 and SN2X pathways depends on multiple factors:
- Leaving Group Characteristics: S_N2X reactions appear particularly favorable with polarizable halogens that can participate in halogen bonding interactions [36]
- Nucleophile Properties: Nucleophiles capable of engaging in halogen bonding may favor the S_N2X pathway [36]
- Catalyst Effects: Cationic catalysts can promote S_N2X reactions by stabilizing developing charges in the transition state [36]
- Substrate Structure: While traditional SN2 reactions favor primary alkyl centers, SN2X demonstrates distinct substrate preferences that continue to be elucidated [4]
Research Reagent Solutions for Pathway Investigation
Table 3: Essential Research Reagents for Studying S_N2 and S_N2X Mechanisms
| Reagent Category | Specific Examples | Function in Research | Mechanistic Relevance |
|---|---|---|---|
| Standard Leaving Groups | Iodide (I⁻), Bromide (Br⁻), Chloride (Cl⁻), Tosylate (OTs), Triflate (OTf) | Establishing baseline reactivity and rate comparisons | S_N2 benchmarking [78] [77] |
| Stereochemical Probes | Enantiomerically pure alkyl halides, chiral stationary phase HPLC columns, polarimeters | Determining stereochemical course (inversion vs. retention) | Distinguishing SN2 vs SN2X [4] [80] |
| Halogen Bonding Nucleophiles | Thiocresolates, azide (N₃⁻), cyanide (CN⁻) | Promoting frontside attack pathways | S_N2X pathway investigation [36] [77] |
| Cationic Catalysts | Designed organocatalysts with cationic centers | Stabilizing transition states for frontside attack | S_N2X enantioconvergence [36] |
| Computational Tools | Quantum chemistry software (Gaussian, ORCA), kinetic simulation packages | Mapping potential energy surfaces, modeling reaction dynamics | Mechanism validation [4] [9] |
Implications for Pharmaceutical Research
The emergence of the S_N2X pathway presents significant opportunities for pharmaceutical development. The ability to achieve enantioconvergent substitution from racemic starting materials offers efficient routes to enantiomerically enriched building blocks [36]. This is particularly valuable in drug development where different enantiomers often exhibit distinct pharmacological profiles, metabolic fates, and toxicological properties.
Furthermore, the SN2X mechanism expands the synthetic toolbox available to medicinal chemists, enabling novel disconnection strategies that may provide more efficient access to complex molecular architectures. The halogen bonding interactions that facilitate SN2X reactions may also have implications for molecular recognition in biological systems, potentially informing drug design strategies targeting halogen-rich binding sites.
Leaving group ability represents a fundamental molecular property with profound implications for reaction pathway selection in nucleophilic substitution chemistry. While traditional SN2 reactions follow well-established principles with backside attack and inversion of configuration, the recently characterized SN2X mechanism demonstrates that alternative pathways involving frontside attack and halogen bonding interactions can compete under specific conditions. The quantitative assessment of leaving group competence, combined with sophisticated kinetic analyses and computational studies, provides researchers with powerful tools to understand and manipulate these competing pathways. For pharmaceutical scientists and synthetic chemists, mastery of these principles enables strategic control of reaction outcomes and stereochemistry, facilitating more efficient synthesis of complex molecules with defined stereochemical properties.
Nucleophilic substitution reactions at carbon centers, the archetypal SN2 mechanism, are characterized by a double-well potential energy surface (PES) and a concerted backside attack that results in inversion of configuration. However, this paradigm shifts fundamentally when the central atom is a higher-period element such as silicon, germanium, or phosphorus. This technical guide examines the distinct mechanistic pathways for nucleophilic substitution at these heteroatoms, where the PES transitions to a single-well profile and reactions may proceed through stable pentacoordinate intermediates. We explore the experimental and computational evidence supporting these mechanisms, provide detailed protocols for their study, and discuss implications for research in chemical synthesis and drug development.
The classic SN2 reaction at a carbon center follows a concerted mechanism with a single transition state and a double-well PES, representing the reactant and product complexes [9]. This pathway proceeds exclusively via backside attack, resulting in stereospecific inversion of configuration at the chiral center [1] [3]. The reaction rate follows second-order kinetics, dependent on both nucleophile and substrate concentrations [54] [59].
When the central atom extends beyond the second period to elements like silicon, germanium, and phosphorus, a profound mechanistic shift occurs. The expanded valence shell and larger atomic radius of these elements enable the formation of stable pentacoordinate intermediates, fundamentally altering the reaction pathway [9] [82]. This transition from a double-well to single-well PES represents a cornerstone distinction in substitution chemistry at heteroatoms, with significant implications for reaction design in synthetic and medicinal chemistry.
Mechanistic Foundations and Potential Energy Surfaces
Comparative Potential Energy Surfaces
Table 1: Comparison of Potential Energy Surfaces for Nucleophilic Substitution
| Feature | SN2@Carbon | SN2@Silicon/Germanium | SN2@Phosphorus |
|---|---|---|---|
| PES Type | Double-well | Single-well | Triple-well (for stepwise) |
| Intermediate | None | Stable pentacoordinate | Trigonal bipyramidal intermediate (TBI) |
| Charge Transfer | Abrupt near TS | Gradual | Depends on mechanism |
| Solvent Sensitivity | High | Low | Variable |
| Stereochemical Outcome | Inversion | Inversion (via backside attack) | Inversion (via A–E mechanism) |
Key Mechanistic Pathways
Diagram 1: Comparative substitution pathways for different central atoms
Experimental Evidence and Case Studies
Identity Substitution Reactions at Phosphorus
Green and Hudson (1963) conducted pioneering work on identity methoxy exchange in methyl ethylphenylphosphinate, providing early evidence for the distinct mechanism at phosphorus centers [82]. Their experimental protocol involved:
Experimental Protocol 1: Identity Methoxy Exchange at Phosphorus
- Substrate Preparation: Optically active methyl ethylphenylphosphinate was synthesized with confirmed enantiomeric purity
- Isotopic Labeling: A separate batch of substrate was prepared with 14C-labeled methoxy group for exchange rate studies
- Kinetic Measurements:
- Racemization rate (krac) determined using polarimetry with optically active substrate
- Exchange rate (kexch) measured using radiochemical techniques with 14C-labeled substrate
- Reactions conducted in methanol solvent with methoxide ion as nucleophile
- Temperature Control: Experiments performed under isothermal conditions (temperature not specified in source)
- Data Analysis: Comparison of krac and kexch to determine stereochemical outcome
The key finding was that krac = 2 × kexch, demonstrating that each substitution event proceeds with inversion of configuration, consistent with a stereospecific mechanism [82].
Computational Studies of Reaction Pathways
Density functional theory (DFT) calculations have provided atomic-level insight into these mechanisms. For the identity methoxy exchange at phosphorus:
Computational Protocol 1: DFT Analysis of Phosphorus Substitution
- Method Selection: Employ density functional theory with appropriate functional (e.g., B3LYP) and basis sets
- Geometry Optimization: Locate all stationary points on the potential energy surface:
- Reactant complex (RC)
- Transition states (TS1, TS2)
- Trigonal bipyramidal intermediate (TBI)
- Product complex (PC)
- Frequency Calculations: Verify nature of stationary points (minima for RC, TBI, PC; first-order saddle points for TS1, TS2)
- Intrinsic Reaction Coordinate: Follow IRC paths from transition states to connected minima
- Energy Calculations: Compute relative free energies for all species
- Bond Analysis: Examine bond lengths and angles along reaction path
DFT calculations revealed that this identity reaction proceeds through a stepwise addition-elimination (A-E) mechanism featuring a stable trigonal bipyramidal phosphorus intermediate (TBI-1) with two methoxy groups in apical positions [82].
Table 2: Experimental Kinetic Data for Identity Substitution Reactions
| Reaction System | Central Atom | krac/kexch Ratio | Mechanism Determined |
|---|---|---|---|
| Methyl ethylphenylphosphinate + MeO⁻ | Phosphorus | 2.0 | Stepwise A–E with TBI |
| (Ethoxy)ethylphosphonochloridothionate + Cl⁻ | Phosphorus | 2.0 | Concerted SN2-P |
| Methyl p-toluenesulfinate + MeO⁻ (acid cat.) | Sulfur | 2.0 | Stepwise A–E with sulfurane intermediate |
| sec-Octyl iodide + I⁻ | Carbon | 2.0 | Concerted SN2 |
Silicon and Germanium Centers
For silicon and germanium centers, the larger atomic radius and more diffuse orbitals lead to a single-well PES with a stable pentacoordinate intermediate [9]. The key distinctions include:
- Gradual Charge Transfer: Unlike the abrupt charge transfer in carbon SN2 reactions, silicon and germanium exhibit more gradual charge transfer due to their larger LUMOs [9]
- Reduced Solvent Effects: The PES curvature for silicon and arsenic centers is more resistant to solvent effects compared to carbon and phosphorus [9]
- Steric Considerations: Despite the stable intermediate, backside attack remains preferred due to orbital alignment requirements
Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Heteroatom Substitution
| Reagent/Category | Example Specific Compounds | Function in Research |
|---|---|---|
| Phosphinate Esters | Methyl ethylphenylphosphinate | Model substrate for studying phosphorus substitution mechanisms |
| Isotopically Labeled Reagents | 14C-methanol, radioactive iodide ( *I⁻ ) | Tracing exchange rates in identity substitution reactions |
| Nucleophiles | Methoxide ion (MeO⁻), chloride (Cl⁻) | Attacking species in substitution studies |
| Polar Aprotic Solvents | Acetone, DMSO, DMF | Reaction medium for minimizing solvation effects on nucleophiles |
| Computational Tools | DFT software packages | Theoretical studies of potential energy surfaces and reaction pathways |
| Acid Catalysts | Trifluoroacetic acid | Facilitating substitution at sulfur centers in sulfinate esters |
Methodologies for Mechanistic Distinction
Kinetic Analysis Protocol
Protocol 2: Distinguishing Concerted vs. Stepwise Mechanisms
Stereochemical Analysis:
- Prepare optically active substrate
- Measure racemization rate (k_rac) using polarimetry or chiral chromatography
- Measure exchange rate (k_exch) using isotopic labeling
- Calculate krac/kexch ratio
Interpretation:
- krac/kexch = 2: Stereospecific inversion (consistent with either concerted SN2 or stepwise A-E without pseudorotation)
- krac/kexch = 1: Racemization (suggesting stable intermediate with pseudorotation)
Computational Verification:
- Perform DFT calculations to locate intermediates
- Calculate energy barriers for pseudorotation vs. direct decomposition
- Construct complete potential energy surface
Computational Workflow
Diagram 2: Computational workflow for mechanism determination
Implications for Research and Development
The mechanistic distinctions in nucleophilic substitution at heteroatoms have significant implications:
- Catalyst Design: Understanding pentacoordinate intermediates at phosphorus enables design of phosphorus-based catalysts for organic synthesis
- Materials Science: Substitution at silicon centers informs silicone polymer synthesis and surface functionalization strategies
- Drug Development: Phosphorus-containing compounds are prevalent in pharmaceuticals (e.g., nucleotide analogs, protease inhibitors); understanding their reactivity aids drug design
- Synthetic Methodology: Knowledge of heteroatom substitution mechanisms enables development of new synthetic routes to organosilicon and organophosphorus compounds
Nucleophilic substitution at silicon, germanium, and phosphorus centers follows fundamentally different pathways compared to classic carbon-centered SN2 reactions. The expanded valence capabilities of these higher-period elements enable the formation of stable pentacoordinate intermediates, leading to single-well or triple-well potential energy surfaces distinct from the double-well PES of carbon systems. Experimental techniques combining kinetic studies with stereochemical analysis, complemented by modern computational methods, provide powerful tools for elucidating these mechanisms. This understanding enables researchers to harness the unique reactivity of heteroatoms for applications spanning synthetic chemistry, materials science, and drug development.
The bimolecular nucleophilic substitution (S_N2) reaction is a cornerstone of organic chemistry, characterized by a concerted mechanism in which the formation of the new nucleophile-carbon bond and the breaking of the carbon-leaving group bond occur simultaneously via a backside attack [3] [83]. This pathway proceeds through a single transition state with trigonal bipyramidal geometry, inevitably resulting in inversion of stereochemistry at the chiral center [3] [63]. The requirement for backside attack stems from the need for the nucleophile to access the σ* antibonding orbital of the carbon-leaving group bond, which is oriented 180 degrees from the bond itself [3].
While this description represents the archetypal S_N2 mechanism, emerging research reveals that specific structural factors can disrupt this conventional pathway and potentially enable alternative attack trajectories. This whitepaper examines the structural constraints that govern nucleophilic approach and explores exceptional molecular architectures where these constraints are circumvented, potentially allowing for novel substitution pathways including frontside mechanisms. Understanding these exception cases holds significant implications for reaction design in complex molecule synthesis, particularly in pharmaceutical development where stereochemical outcomes critically determine biological activity.
Conventional S_N2 Mechanism and Steric Limitations
The Backside Attack Paradigm
The classical S_N2 mechanism mandates nucleophilic approach from the side opposite the leaving group, resulting in a well-defined transition state with characteristic features [3]:
- Concerted bond formation/cleavage: The nucleophile attacks as the leaving group departs
- Stereochemical inversion: The reaction proceeds with Walden inversion
- Second-order kinetics: Rate = k[substrate][nucleophile]
- Transition state geometry: Trigonal bipyramidal with partial bonds to nucleophile and leaving group
This backside attack occurs 180° to the C-Leaving Group bond because the nucleophile must donate electrons into the antibonding (σ*) orbital of this bond, which resides directly opposite the bond itself [3]. The resulting steric requirements for this approach are stringent, making the reaction highly sensitive to substitution patterns at the electrophilic carbon.
Steric Effects on Reaction Rates
The reactivity of alkyl halides in conventional S_N2 reactions follows a predictable pattern based on steric hindrance around the electrophilic carbon. As substitution increases, the nucleophile experiences greater steric repulsion when attempting the required backside approach, dramatically reducing reaction rates [84].
Table 1: Relative Rates of S_N2 Reactions Based on Substrate Structure
| Substrate Type | Example | Relative Rate | Structural Rationale |
|---|---|---|---|
| Methyl | CH₃-Br | ~30 | Minimal steric hindrance to backside approach |
| Primary | CH₃CH₂-Br | 1 | Reference compound |
| Secondary | (CH₃)₂CH-Br | 0.02 | Significant steric shielding |
| Tertiary | (CH₃)₃C-Br | ~0 | Complete blockage of backside approach |
Beyond α-substitution, β-branching further impedes the SN2 reaction. For example, 2-methyl-1-bromopropane reacts significantly slower than 1-bromopropane due to steric interference from the adjacent methyl group [84]. This exquisite sensitivity to molecular architecture normally precludes SN2 reactivity for highly substituted substrates, yet certain structural modifications can circumvent these limitations.
Structural Factors Enabling Alternative Pathways
Electrophilic Center Modification
While carbon represents the canonical electrophilic center in S_N2 reactions, substitution with higher-period elements fundamentally alters the reaction potential energy surface (PES). Computational studies reveal that changing the central atom from a second-period element (e.g., carbon) to a higher-period element (e.g., silicon, germanium) transitions the PES from a double-well to a single-well profile [9].
Table 2: Effect of Central Atom Identity on S_N2 Potential Energy Surface
| Central Atom | Period | PES Type | Key Characteristics | Implications |
|---|---|---|---|---|
| Carbon | 2 | Double-well | Distinct reactant and product complexes | Conventional two-step S_N2 with inversion |
| Silicon | 3 | Single-well | No intermediate complexes | Possible alternative mechanisms |
| Germanium | 4 | Single-well | Shallow or non-existent minima | Lower stereoselectivity potential |
This transition occurs because larger central atoms have more diffuse orbitals, permitting more gradual charge transfer and reduced steric constraints in the transition state [9]. The resulting single-well PES for silicon and germanium systems indicates a fundamentally different reaction coordinate that may accommodate alternative nucleophilic approaches.
Steric Shielding and Molecular Architecture
Constrained molecular architectures can physically block the conventional backside approach while potentially creating new trajectories for nucleophilic attack. The classic example comes from bridgehead systems such as 1-bromotriptycene, where the bicyclic structure completely prevents backside attack while simultaneously disfavoring carbocation formation due to bridgehead geometry constraints [3]. Such systems exhibit remarkable inertness toward nucleophilic substitution under standard conditions.
However, carefully designed substrates with strategic steric protection of the backside approach while maintaining accessibility to alternative trajectories may enable previously inaccessible mechanisms. Computational models suggest that bulky substituents can be strategically employed not merely to inhibit reactivity, but to redirect it along energetically accessible frontside pathways in specifically engineered systems.
Experimental Methodologies for Investigating Alternative Pathways
Kinetic Analysis Techniques
Establishing reaction mechanisms requires rigorous kinetic analysis. For investigating potential alternative pathways, the following methodologies are essential:
Variable-Time Kinetic Monitoring
- Prepare substrate solutions at multiple concentrations (typically 0.01-0.1M)
- Introduce nucleophile in at least 10-fold excess to maintain pseudo-first-order conditions
- Monitor reaction progress via HPLC/GC sampling at t=0, 5, 10, 15, 30, 60, 120 minutes
- Plot ln([substrate]₀/[substrate]ₜ) vs time; linear plot indicates first-order kinetics in substrate
- Repeat across nucleophile concentrations; slope dependence indicates reaction order
Activation Parameter Determination
- Conduct kinetic measurements at minimum five temperatures (typically 0°C, 25°C, 50°C, 75°C, 100°C)
- Apply Eyring equation: ln(k/T) = -ΔH‡/RT + ΔS‡/R + ln(kB/h)
- Plot ln(k/T) vs 1/T to determine ΔH‡ (enthalpy) and ΔS‡ (entropy)
- Highly negative ΔS‡ suggests ordered transition state (characteristic of conventional S_N2)
Stereochemical Analysis
Determining stereochemical outcomes provides critical evidence for mechanistic pathways:
Chiral Substrate Synthesis
- Prepare enantiomerically pure substrates using established asymmetric methodologies
- Employ chiral auxiliary approaches or enzymatic resolutions
- Verify enantiopurity by chiral HPLC or optical rotation measurements
Stereochemical Outcome Analysis
- Monitor racemization vs inversion using chiral stationary phase HPLC
- Employ polarimetry for rapid assessment of stereochemical changes
- For proposed frontside pathways, analyze products for unexpected retention patterns
Computational Modeling Approaches
Modern computational chemistry provides atomic-level insight into alternative reaction pathways:
Potential Energy Surface Mapping
- Employ density functional theory (e.g., ωB97X-D/def2-TZVP) for geometry optimizations
- Conduct relaxed surface scans along reaction coordinate
- Identify stationary points (minima, transition states) via frequency calculations
- Apply solvation models (e.g., CPCM, SMD) to account for solvent effects
Activation Strain Analysis
- Decompose activation energies into strain and interaction components
- Quantify steric and electronic contributions to reaction barriers
- Identify structural features that stabilize alternative transition states
Figure 1: Experimental Workflow for Mechanism Elucidation
Research Reagent Solutions for Mechanistic Studies
Table 3: Essential Research Reagents for Investigating Alternative S_N2 Pathways
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Sterically-Hindered Substrates | 1-Bromotriptycene, Neopentyl halides, 2,2,6,6-Tetramethylpiperidine derivatives | Probe steric limits of conventional mechanism | Bridgehead systems test backside accessibility; neopentyl systems reveal β-branching effects |
| Alternative Electrophilic Centers | Chlorosilanes, Germanium halides, Organotin halides | Investigate period-dependent mechanistic changes | Larger atomic radii alter orbital symmetry and steric requirements |
| Bulky Nucleophiles | Potassium tert-butoxide, DABCO, 2,6-Di-tert-butylpyridine | Assess nucleophile size requirements | Differentiate between steric and electronic effects |
| Solvent Systems | Ionic liquids, [BMIM][BF₄], Diglyme, Acetonitrile, DMSO | Modulate solvent polarity and ion pairing | Polar aprotic solvents enhance nucleophilicity; ionic liquids can dramatically alter kinetics [63] |
| Leaving Group Modifiers | Triflates, Tosylates, Mesylates, Lewis acids (BF₃, AlCl₃) | Enhance poor leaving groups | Triflate, tosylate, mesylate anions stabilize incipient negative charge [63]; Lewis acids activate hydroxyl groups |
The structural factors governing S_N2 reactivity extend beyond simple steric bulk to encompass atomic period, orbital symmetry, and three-dimensional architecture. While the conventional backside attack mechanism remains dominant for standard substrates, strategic molecular design can create exception cases that enable alternative pathways. The most promising avenues for future research include:
Precision Steric Engineering - Designing substrates with calculated steric protection of the backside approach while maintaining controlled accessibility to alternative trajectories. Such systems would test the limits of frontside attack feasibility.
Period-Engineered Electrophiles - Systematic investigation of higher-period central atoms (Si, Ge, Sn) to exploit their altered potential energy surfaces and potentially lower stereoelectronic requirements.
Computational Prediction - Developing machine learning models to predict when structural modifications will redirect reactivity rather than simply inhibiting it.
These research directions hold particular promise for pharmaceutical development, where controlled stereochemical outcomes in sterically congested molecular environments remain a significant synthetic challenge. By understanding and exploiting these exception cases, medicinal chemists may gain new strategies for constructing complex stereochemical architectures that evade conventional synthetic approaches.
Mechanistic Validation and Comparative Analysis of Substitution Pathways
Nucleophilic substitution reactions represent a cornerstone of organic chemistry, with the SN1 (substitution nucleophilic unimolecular) and SN2 (substitution nucleophilic bimolecular) mechanisms governing fundamental molecular transformations. These reactions follow distinctly different pathways, leading to profound differences in their stereochemical outcomes. While traditional understanding has centered on these two principal mechanisms, recent research has uncovered additional pathways such as the halogenophilic nucleophilic substitution (SN2X) reaction, which exhibits a distinctly different reaction pathway compared to the conventional SN2 mechanism yet can lead to the same reaction products [4] [5]. This technical guide provides an in-depth analysis of these mechanisms, emphasizing their stereochemical consequences and the experimental methodologies used to distinguish between them, with particular relevance to drug development where stereochemical purity often determines therapeutic efficacy and safety.
The critical distinction between SN1 and SN2 mechanisms lies in their molecular-level pathways. The SN2 mechanism proceeds through a single, concerted step where bond formation and bond breakage occur simultaneously, while the SN1 mechanism occurs through a stepwise process involving a carbocation intermediate [61] [85]. This fundamental mechanistic difference dictates all subsequent stereochemical outcomes and forms the basis for predicting and controlling the stereochemistry of substitution products—a crucial consideration in pharmaceutical synthesis where specific stereoisomers may possess dramatically different biological activities.
Mechanistic Pathways of Nucleophilic Substitution
The SN2 Mechanism: Concerted Backside Attack
The SN2 mechanism is characterized by a single, concerted step in which the nucleophile attacks the electrophilic carbon center from the backside relative to the leaving group, while the leaving group departs simultaneously. This synchronous process occurs through a pentacoordinated transition state where the central carbon temporarily adopts a trigonal bipyramidal geometry [61] [85]. The reaction is termed "bimolecular" because the rate-determining step involves two molecular entities: the nucleophile and the substrate.
The backside attack is sterically demanding, requiring direct access to the electrophilic carbon. Consequently, the SN2 reaction is highly sensitive to steric hindrance around the reaction center, proceeding most readily with methyl and primary alkyl halides, slower with secondary substrates, and essentially non-existent with tertiary alkyl halides [61] [86]. The stereochemical consequence of this concerted backside attack is an inversion of configuration at the carbon center, analogous to an umbrella turning inside out in the wind [61] [85].
The SN1 Mechanism: Stepwise Carbocation Formation
In contrast to the concerted SN2 pathway, the SN1 mechanism proceeds through a stepwise process involving two critical steps. The initial and rate-determining step is the unimolecular dissociation of the carbon-leaving group bond to form a planar carbocation intermediate. This carbocation possesses sp² hybridization and trigonal planar geometry, with an empty p orbital extending above and below the molecular plane [61] [87] [85].
The second step involves nucleophilic attack on this carbocation intermediate. Since the carbocation is planar and achiral, the nucleophile can approach with equal probability from either face of the plane, leading to the formation of both possible stereoisomers [87] [85]. This loss of stereochemical integrity results in racemization when substitution occurs at a chiral center, though complete 50:50 racemization is rarely observed in practice due to various shielding effects [87].
Table 1: Fundamental Characteristics of SN1 and SN2 Mechanisms
| Characteristic | SN1 Mechanism | SN2 Mechanism |
|---|---|---|
| Molecularity | Unimolecular (rate-determining step) | Bimolecular |
| Reaction Steps | Two or more steps | Single concerted step |
| Intermediate | Carbocation | None (transition state only) |
| Rate Law | Rate = k[substrate] | Rate = k[substrate][nucleophile] |
| Rate Determining Step | Carbocation formation | Backside attack transition state |
Emerging Mechanisms: SN2X and SN2Ch Pathways
Recent research has identified additional nucleophilic substitution pathways that expand our understanding beyond the traditional SN1/SN2 dichotomy. The halogenophilic nucleophilic substitution (SN2X) mechanism follows a distinctly different pathway from classical SN2 reactions, though both can yield identical products [4] [5]. In SN2X reactions, the nucleophile initially attacks the halogen atom (the leaving group) rather than the carbon center, forming a hypervalent halogen intermediate before the leaving group departs.
Quantitative studies of SN2X reactions have revealed the presence of a pro-chiral anion intermediate, contrasting with the stereospecific nature of classical SN2 reactions [4]. Researchers have developed kinetic simulation procedures to measure the halogenophilic percentage (X%) and established a new parameter—relative halogenophilicity (H)—to quantify the intrinsic characteristics of SN2X reactions [5]. This parameter correlates well with established physical organic chemistry principles such as the Hammett and Mayr postulates [5].
Further investigations have identified additional mechanisms including chalcogenophilic nucleophilic substitution (SN2Ch) and bromide-catalyzed dynamic kinetic resolution [4]. These pathways exhibit similar behavior from both thermodynamic and kinetic perspectives, suggesting they occur to varying degrees in most substitution reactions and should be considered as interconnected rather than isolated phenomena [5].
Stereochemical Outcomes and Consequences
SN2: Inversion of Configuration
The stereochemical outcome of the SN2 reaction is unequivocal: complete inversion of configuration at the reaction center. This stereospecificity arises from the mandatory backside attack mechanism, which ensures that the nucleophile always enters opposite to the departing leaving group [61] [85]. The three substituents not involved in the reaction flip their spatial orientation during the transition from the starting material to the product, much like an umbrella turning inside out in strong wind [85].
This inversion phenomenon, known as the Walden inversion, has profound implications in asymmetric synthesis. When a chiral secondary alkyl halide undergoes SN2 substitution, the product displays a configuration opposite to that of the starting material [85]. This predictable stereochemical outcome makes the SN2 mechanism particularly valuable in synthetic planning where specific stereochemistry is required.
SN1: Racemization with Partial Retention
The stereochemical consequence of the SN1 mechanism is racemization, resulting from the planar, achiral nature of the carbocation intermediate [87] [85]. Since the nucleophile can approach with equal probability from either face of the flat carbocation, the reaction typically produces a racemic mixture containing equal amounts of enantiomers when starting from a single enantiomer [85].
However, complete 50:50 racemization is rarely observed in practice. Often, the reaction displays partial retention of configuration due to the leaving group not fully dissociating from the reaction sphere before nucleophilic attack occurs [87]. In such instances, the departing leaving group can partially block one face of the carbocation, leading to uneven stereochemical outcomes. This phenomenon, known as ion pairing, results in the original leaving group shielding one face of the carbocation and leading to a preference for the nucleophile attacking from the opposite side, yielding more inversion than retention products [87].
Stereochemical Pathways in SN1 and SN2 Reactions
Table 2: Comparative Stereochemical Outcomes of SN1 and SN2 Reactions
| Stereochemical Aspect | SN1 Mechanism | SN2 Mechanism |
|---|---|---|
| Stereochemical Outcome | Racemization (with possible partial retention) | Complete inversion of configuration |
| Intermediate Geometry | Planar carbocation (sp² hybridized) | Trigonal bipyramidal transition state |
| Stereospecificity | Non-stereospecific | Stereospecific |
| Nucleophilic Attack | Equally probable from both faces | Exclusive backside attack only |
| Effect of Ion Pairing | Can lead to uneven distribution of enantiomers | Minimal effect |
Experimental Determination and Methodologies
Kinetic Analysis Techniques
Determining the operative mechanism for a nucleophilic substitution reaction begins with kinetic analysis. The fundamental distinction lies in the rate law: SN1 reactions exhibit first-order kinetics (rate = k[substrate]), while SN2 reactions display second-order kinetics (rate = k[substrate][nucleophile]) [61] [85] [88].
Experimentally, this is determined by measuring reaction rates under varying concentrations of nucleophile and substrate. For SN1 reactions, doubling the substrate concentration doubles the reaction rate, while changing nucleophile concentration has no effect on the rate [61] [85]. Conversely, for SN2 reactions, doubling either substrate or nucleophile concentration doubles the reaction rate, while doubling both quadruples the rate [85]. These kinetic studies are typically conducted using techniques such as UV-Vis spectroscopy, NMR spectroscopy, or chromatography to monitor reaction progress under controlled conditions.
Stereochemical Analysis Methods
Stereochemical analysis provides definitive evidence for distinguishing between SN1 and SN2 mechanisms. When a reaction occurs at a chiral center, the configuration of the product reveals the operative mechanism [61] [87].
For SN2 reactions occurring at chiral centers, stereochemical analysis confirms inversion of configuration. This is typically demonstrated using enantiomerically pure substrates and analyzing the products through polarimetry, chiral chromatography, or NMR with chiral shift reagents [85]. The specific rotation of the product should be equal in magnitude but opposite in sign to the starting material when the SN2 mechanism operates exclusively.
For SN1 reactions, stereochemical analysis reveals racemization. The product mixture shows diminished optical activity, with the specific rotation approaching zero as the reaction proceeds to completion [87] [85]. Modern analytical techniques including chiral HPLC, GC with chiral stationary phases, and NMR spectroscopy with chiral solvating agents provide precise enantiomeric ratios that confirm the extent of racemization [87].
Quantitative Studies of Competing Pathways
Advanced research methodologies now enable quantitative analysis of competing substitution pathways. Recent studies have developed procedures supported by kinetic simulations to measure the halogenophilic percentage (X%) in reactions where both SN2 and SN2X pathways operate simultaneously [4] [5].
These methodologies typically involve stereospecific substrates where the distinct stereochemical outcomes of different pathways can be tracked. By monitoring the time dependency of product ratios under varied reaction conditions, researchers can quantify the contribution of each mechanism [5]. These sophisticated approaches have led to the discovery of additional mechanisms such as chalcogenophilic nucleophilic substitution (SN2Ch) and bromide-catalyzed dynamic kinetic resolution [4].
Experimental Workflow for Mechanism Determination
Research Reagent Solutions for Mechanistic Studies
Table 3: Essential Research Reagents for Nucleophilic Substitution Studies
| Reagent Category | Specific Examples | Research Application | Function in Mechanistic Studies |
|---|---|---|---|
| Alkyl Halide Substrates | Tert-butyl bromide, Bromoethane, (S)-2-Bromooctane | SN1 vs. SN2 determination | Tertiary substrates favor SN1; primary substrates favor SN2; chiral substrates for stereochemical analysis |
| Nucleophiles | NaCN, KOEt, CH₃OH, H₂O | Nucleophile strength studies | Strong nucleophiles (CN⁻) favor SN2; weak nucleophiles (H₂O) favor SN1 |
| Solvents | Water, Ethanol, Acetone, DMSO | Solvent effect studies | Polar protic solvents (H₂O) favor SN1; polar aprotic solvents (DMSO) favor SN2 |
| Analytical Reagents | Chiral shift reagents, Chiral HPLC columns | Stereochemical analysis | Determine enantiomeric excess and distinguish between inversion and racemization |
| Kinetic Probes | Isotopically labeled compounds, UV-active substrates | Kinetic studies | Track reaction rates and determine rate laws |
Factors Governing Mechanism Selection
Substrate Structure Effects
The structure of the electrophilic substrate is the most significant factor in determining whether an SN1 or SN2 pathway will dominate. This structural dependence arises from the fundamentally different requirements of the two mechanisms [61] [86] [88].
For SN2 reactions, steric accessibility of the backside of the carbon-leaving group bond is crucial. Methyl and primary alkyl halides react rapidly via the SN2 mechanism due to minimal steric hindrance. Secondary alkyl halides react more slowly, while tertiary alkyl halides are essentially unreactive due to steric blocking of the required backside approach [61] [88].
For SN1 reactions, the stability of the carbocation intermediate determines reactivity. Tertiary alkyl halides react rapidly via the SN1 mechanism because they form relatively stable tertiary carbocations. Secondary alkyl halides react more slowly, while primary and methyl carbocations are so unstable that SN1 reactions are not observed under normal conditions [61] [86]. The order of reactivity is precisely opposite for the two mechanisms: tertiary > secondary > primary for SN1, and primary > secondary > tertiary for SN2 [61] [88].
Nucleophile and Solvent Effects
The nature of the nucleophile and solvent profoundly influences the competition between SN1 and SN2 pathways. SN2 reactions require strong nucleophiles, typically anions or other electron-rich species with high chemical reactivity [86] [88]. The nucleophile must be sufficiently reactive to directly displace the leaving group in a single concerted step. In contrast, SN1 reactions involve weak nucleophiles, often the solvent itself, since the rate-determining step does not involve nucleophilic participation [86] [88].
Solvent effects further distinguish the two mechanisms. SN2 reactions are favored by polar aprotic solvents such as acetone, DMSO, and acetonitrile, which solvate cations strongly but leave anions relatively naked and highly reactive [88]. SN1 reactions are favored by polar protic solvents such as water and alcohols, which stabilize the carbocation intermediate and the leaving group through solvation and hydrogen bonding [87] [88]. These solvents effectively lower the activation energy for the rate-determining ionization step through solvation of the developing charges.
Table 4: Comprehensive Comparison of SN1 and SN2 Reaction Parameters
| Reaction Parameter | SN1 Mechanism | SN2 Mechanism |
|---|---|---|
| Alkyl Halide Structure | Tertiary > Secondary > Primary | Primary > Secondary > Tertiary |
| Nucleophile | Weak (often solvent) | Strong |
| Solvent | Polar protic (e.g., H₂O, ROH) | Polar aprotic (e.g., DMSO, acetone) |
| Kinetics | First-order | Second-order |
| Stereochemistry | Racemization | Inversion |
| Rate Equation | Rate = k[substrate] | Rate = k[substrate][nucleophile] |
| Reactive Intermediate | Carbocation | Transition state only |
| Effect of [Nu] on Rate | No effect | Direct proportionality |
| Leaving Group Importance | Critical (affects carbocation formation) | Critical (affects transition state stability) |
Implications for Drug Development and Future Research
The stereochemical outcomes of nucleophilic substitution reactions have profound implications in pharmaceutical research and development, where precise stereochemical control is often essential for drug efficacy and safety. Understanding the factors that govern SN1 versus SN2 pathways enables medicinal chemists to design synthetic routes that deliver the correct stereoisomer of drug candidates [89].
Recent advances in understanding alternative mechanisms like SN2X and SN2Ch open new possibilities for controlling stereochemistry in complex synthetic targets [4] [5]. The discovery that these pathways are inherently linked and occur to varying degrees in most substitution reactions suggests a more nuanced approach to reaction design is necessary [5]. The development of quantitative parameters like relative halogenophilicity (H) provides researchers with new tools to predict and control these complex reaction networks [5].
Future research directions will likely focus on expanding our understanding of these interconnected reaction pathways and developing catalytic systems that can steer reactions toward desired stereochemical outcomes. The integration of kinetic simulations with experimental data, as demonstrated in recent SN2X studies, represents a powerful approach to deconvoluting complex reaction mechanisms [4]. As these methodologies advance, they will enable more precise stereochemical control in pharmaceutical synthesis, ultimately contributing to the development of more effective and safer therapeutic agents.
Nucleophilic substitution stands as a foundational reaction class in organic chemistry with profound implications for synthetic methodology and biochemical process elucidation. This technical guide provides a comprehensive transition state analysis comparing the two principal mechanistic pathways: the concerted backside attack mechanism (SN2) and the stepwise mechanism involving carbocation intermediates (SN1). Within the context of advanced SN2X reaction mechanism research, understanding these fundamental pathways provides the essential framework for exploring more complex concerted and stepwise substitutions. The distinct transition states and reactive intermediates involved dictate divergent kinetic behaviors, stereochemical outcomes, and substrate selectivity, each critical for researchers designing synthetic routes or investigating biomolecular nucleophilic reactions in drug development.
Mechanistic Pathways and Transition States
The SN2 Concerted Backside Attack Mechanism
The SN2 mechanism proceeds via a single, concerted step wherein nucleophilic attack and leaving group departure occur synchronously through a single transition state [61]. This bimolecular nucleophilic substitution features a characteristic backside attack where the nucleophile approaches the electrophilic carbon from a trajectory 180° opposite the leaving group (C–LG bond) [3]. This trajectory maximizes overlap between the nucleophile's lone pair and the σ* antibonding orbital of the C–LG bond while minimizing steric and electronic repulsion [1].
The transition state exhibits a trigonal bipyramidal geometry where the nucleophile and leaving group occupy the apical positions with partial bond character [61]. This five-coordinate carbon transition state exists transiently before collapsing to products with inversion of configuration at the reaction center [3]. The reaction follows second-order kinetics, with rate dependent on both nucleophile and substrate concentrations [61].
Table 1: Key Characteristics of the SN2 Transition State
| Characteristic | Description |
|---|---|
| Molecularity | Bimolecular (2 molecules in rate-determining step) |
| Transition State Geometry | Trigonal bipyramidal |
| Bond Formation/Cleavage | Concerted with partial bonds in TS |
| Stereochemical Outcome | Inversion of configuration |
| Rate Law | Rate = k[Substrate][Nucleophile] |
| Key Orbital Interaction | Nucleophile donates electrons into C–LG σ* orbital |
The SN1 Carbocation-Mediated Mechanism
The SN1 mechanism proceeds through a stepwise pathway involving discrete carbocation intermediates [90]. The reaction initiates with rate-determining heterolysis of the C–LG bond to form a planar, sp²-hybridized carbocation intermediate [61]. This unimolecular dissociation results in a first-order rate dependence solely on substrate concentration [90].
Following carbocation formation, the nucleophile attacks this highly electrophilic intermediate. Since the carbocation possesses trigonal planar geometry with an empty p-orbital, nucleophilic attack can occur with equal probability from either face, resulting in racemization for chiral substrates [61] [90]. The stability of the carbocation intermediate directly governs the reaction rate, with tertiary > secondary > primary > methyl stability trends [91].
Table 2: SN1 Mechanism Intermediates and Transition States
| Species | Geometry | Electron Status | Stability Factors |
|---|---|---|---|
| First Transition State | Dissociative C–LG bond breaking | Partial positive charge developing on carbon | Rate enhanced by electron-donating groups, polar solvents |
| Carbocation Intermediate | Trigonal planar, sp²-hybridized | Empty p-orbital, full positive charge | Tertiary > secondary > primary; stabilized by resonance, hyperconjugation |
| Second Transition State | Associative C–Nu bond formation | Partial bond formation to nucleophile | Faster than first step due to carbocation reactivity |
Quantitative Kinetic and Selectivity Data
Experimental kinetic analyses reveal distinct patterns for SN2 and SN1 mechanisms, particularly regarding substrate preferences and solvent effects. The data below represent relative rate comparisons under standardized conditions for each mechanism.
Table 3: Comparative Reaction Rates for Alkyl Bromides
| Alkyl Bromide | Relative SN2 Rate (with NaCN) | Relative SN1 Rate (in H2O) |
|---|---|---|
| Methyl | 1,250,000 | 1.0 (reference) |
| Ethyl | 1 | 1 |
| Isopropyl | 0.008 | 1,200,000 |
| tert-Butyl | 0.000001 | 1,200,000,000 |
The dramatic rate inversion between SN2 and SN1 pathways highlights the fundamental mechanistic differences: SN2 rates decrease with increasing substitution due to steric hindrance to backside attack, while SN1 rates increase with substitution due to carbocation stabilization through hyperconjugation [61].
Visualization of Mechanistic Pathways
SN2 Backside Attack Mechanism
SN1 Carbocation Mechanism
Experimental Protocols for Mechanism Elucidation
Kinetic Order Determination Protocol
Objective: Determine whether a nucleophilic substitution follows SN1 or SN2 kinetics by measuring reaction order [61] [90].
Materials: Substrate (alkyl halide), nucleophile (e.g., NaI, NaOH), appropriate solvent (polar aprotic for SN2, polar protic for SN1), conductivity meter or GC/HPLC for product quantification.
Procedure:
- Prepare a series of reaction mixtures with constant substrate concentration (e.g., 0.1 M) and varying nucleophile concentrations (0.05 M, 0.1 M, 0.2 M, 0.4 M)
- Monitor reaction progress quantitatively using appropriate analytical method (conductivity, chromatography)
- Determine initial rates from linear portion of concentration-time plots
- Plot log(initial rate) versus log([nucleophile]) - slope of 1 indicates SN2; slope of 0 indicates SN1
- Repeat with varying substrate concentration and constant nucleophile to confirm
Data Interpretation: SN2 shows first-order dependence on both nucleophile and substrate; SN1 shows first-order dependence only on substrate.
Stereochemical Analysis Protocol
Objective: Determine stereochemical outcome to distinguish between inversion (SN2) and racemization (SN1) [61] [1].
Materials: Optically active substrate (e.g., (S)-2-bromooctane), nucleophile, solvent, polarimeter or chiral stationary phase HPLC.
Procedure:
- Dissolve enantiomerically pure substrate in appropriate solvent
- Add nucleophile and allow reaction to proceed to completion
- Isolate product and purify if necessary
- Measure optical rotation or analyze by chiral HPLC
- Calculate enantiomeric excess and percentage racemization/inversion
Data Interpretation: Complete inversion of configuration indicates SN2 pathway; racemic mixture indicates SN1 pathway; partial racemization suggests mixed mechanisms or ion pair effects.
Carbocation Rearrangement Detection Protocol
Objective: Identify carbocation intermediates through detection of rearrangement products characteristic of SN1 mechanism [92].
Materials: Secondary alkyl halide substrate (prone to rearrangement), nucleophile, GC-MS for product identification.
Procedure:
- React substrate under SN1 conditions (polar protic solvent, weak nucleophile)
- Monitor reaction mixture for multiple products
- Isolate and identify products using GC-MS and NMR
- Compare product distribution to possible carbocation rearrangement pathways
Data Interpretation: Presence of products derived from hydride or alkyl shifts confirms carbocation intermediate and SN1 mechanism.
Research Reagent Solutions
Table 4: Essential Research Reagents for Nucleophilic Substitution Studies
| Reagent | Function | Mechanistic Application |
|---|---|---|
| Sodium Iodide in Acetone | Nucleophile source | SN2 probe: I⁻ is excellent nucleophile in polar aprotic solvent |
| Silver Nitrate in Ethanol | Leaving group promoter | SN1 probe: Ag⁺ precipitates halide, driving carbocation formation |
| Deuterated Solvents (CD₃OD, D₂O) | Solvent with kinetic isotope effect | Mechanism tracing via NMR and KIE studies |
| Tosylates/Mesylates | Superior leaving groups | Enhanced reaction rates for both mechanisms |
| Crown Ethers (18-crown-6) | Cation complexation | Enhance nucleophilicity of anions in SN2 by "naked anion" effect |
| Deuterated Alkyl Halides | Substrates with isotope labels | Kinetic isotope effect studies for mechanism elucidation |
Advanced Research Implications
The fundamental understanding of SN2 and SN1 transition states provides critical insights for contemporary research in multiple domains. For SN2X mechanism investigations, the well-characterized orbital interactions and steric requirements of the backside attack mechanism serve as essential reference points for exploring more complex multi-step concerted pathways [93]. The quantitative data on substituent effects and solvent interactions enable computational chemists to refine transition state modeling approaches, particularly important for the development of virtual transition state concepts that average multiple contributing structures [93].
In pharmaceutical development, understanding these mechanistic pathways informs drug design targeting enzyme active sites where nucleophilic substitution occurs, particularly for mechanism-based inhibitors. Recent methodological advances in transition state identification, such as deep learning approaches that treat transition states as out-of-distribution data points in hyperspherical latent space, build directly upon the fundamental principles outlined in this analysis [94]. These computational methods now enable automatic identification of transition states in complex biomolecular systems, accelerating studies of protein dynamics with direct relevance to drug design and biomolecular engineering.
A Technical Whitepaper Framed within SN2X Reaction Mechanism Research
The investigation of reaction mechanisms lies at the heart of predictive organic chemistry and rational drug design. A fundamental dichotomy exists between concerted (simultaneous) and stepwise pathways, where bond formation and cleavage occur in a single kinetic step or through discrete intermediates, respectively [95]. This distinction is not merely academic; it dictates selectivity, stereochemical outcomes, and susceptibility to perturbation, which are critical parameters in synthetic route design and the development of covalent therapeutics.
This whitepaper is framed within a broader thesis on frontside attack nucleophilic substitution, specifically the halogenophilic nucleophilic substitution (SN2X) reaction mechanism [4]. The SN2X pathway represents a fascinating deviation from the classical backside attack SN2 mechanism, involving an initial nucleophilic attack on the halogen (or chalcogen) atom itself rather than the carbon center. This mechanism can compete directly with the traditional SN2 pathway, leading to the same products but through a profoundly different sequence of events involving a pro-chiral anion intermediate [4]. Understanding and quantifying the competition between these simultaneous (concerted SN2) and stepwise (SN2X) pathways is therefore a paradigm for mechanistic elucidation. The tools and concepts discussed herein—kinetic isotope effects, stereochemical analysis, and dynamic trajectory studies—are directly applicable to dissecting the intricate interplay between SN2 and SN2X, providing a framework for the advanced study of nucleophilic substitution landscapes.
Theoretical Framework: The Concerted-Stepwise Continuum
Mechanistic classification is often not binary. The Thornton hypothesis suggests that as a potential intermediate in a stepwise mechanism becomes more stable, the transition state for the corresponding concerted process will geometrically and energetically approach that intermediate [96]. This leads to a continuum where reactions can exist at a "concerted/stepwise boundary," exhibiting characteristics of both. For instance, the C2-C6 cyclization of enyne-allenes features a highly asynchronous transition state where carbon-carbon bond formation significantly precedes hydrogen transfer. Theoretical calculations for this system could not locate a distinct transition state for diradical formation, suggesting the concerted and stepwise pathways merge at the initial transition structure [96]. This blurring of boundaries necessitates sophisticated experimental techniques to map the reaction coordinate and quantify the contribution of competing pathways.
Core Methodologies and Experimental Protocols
Kinetic Isotope Effects (KIE)
Kinetic isotope effects are a powerful tool for probing transition state structure and synchrony. A primary kinetic isotope effect (e.g., kH/kD) arises from changes in zero-point vibrational energy upon isotopic substitution, typically for a bond being broken or formed in the rate-determining step.
Detailed Protocol (Based on Enyne-Allene Cyclization [96]):
- Synthesis of Labeled Substrates: Prepare the substrate of interest with a label (e.g., CD3>) at the position involved in the key transfer step (e.g., hydrogen migration). High isotopic purity (>98%) must be verified via 1H NMR or mass spectrometry.
- Kinetic Monitoring: Conduct separate reactions for labeled and unlabeled substrates under identical, rigorously controlled conditions (solvent, temperature, concentration). The reaction should be followed by a method that quantitatively tracks the disappearance of starting material or appearance of product. In the referenced study, reactions in toluene-d8 at 50°C were monitored by 1H NMR, integrating characteristic aromatic signals against an internal standard [96].
- Data Analysis: Plot the natural logarithm of reactant concentration (or an equivalent measure) versus time. The slope of the linear fit provides the observed first-order rate constant (k). The KIE is the ratio kH/kD.
- Interpretation: A large KIE (>2) indicates significant C-H bond cleavage in the rate-determining transition state, consistent with a concerted mechanism where hydrogen transfer is advanced. A smaller KIE (~1-1.5) suggests a more asynchronous process or a stepwise mechanism where C-H bond cleavage is not central to the initial barrier. The observed KIE of 1.43 for the enyne-allene cyclization indicated a highly asynchronous, borderline mechanism [96].
Stereochemical Analysis and Chirality Probes
The fate of chiral information is a definitive marker for mechanism. This is central to distinguishing SN2 from SN2X pathways [4].
Detailed Protocol for SN2/SN2X Discrimination [4]:
- Chiral Substrate Preparation: Synthesize or obtain an enantiomerically pure alkyl electrophile (e.g., a secondary alkyl halide) with a defined stereocenter at the α-carbon.
- Competition Reaction: Subject the chiral substrate to reaction conditions where both SN2 (inverting) and SN2X pathways are possible. The SN2X mechanism proceeds via a planar, pro-chiral anion intermediate, which can be attacked from either face upon re-aromatization or collapse.
- Product Analysis: Determine the stereochemistry of the product using chiral HPLC, polarimetry, or NMR with chiral shift reagents.
- Quantification: The product distribution reveals the pathway split. Complete inversion implicates a pure SN2 pathway. Racemization or a mixture of stereoisomers indicates involvement of the SN2X pathway (or the SN1 pathway; control experiments are necessary to rule this out). Advanced kinetic simulation of the stereochemical time-course can be used to extract the precise "halogenophilic percentage" (X%) [4].
Computational Dynamics Trajectories
Static quantum mechanical calculations locate minimum energy paths, but they may be inadequate for reactions at the mechanistic boundary. Quasiclassical direct dynamics simulations provide a dynamic view.
Protocol Outline (Informed by [96]):
- Transition State Identification: Use density functional theory (e.g., B3LYP/6-31G(d,p)) to locate the putative concerted transition state.
- Trajectory Initiation: Generate an ensemble of trajectories (e.g., 100+) initialized at the transition state geometry, with atomic momenta assigned based on the vibrational modes at a specific temperature.
- Propagation: Numerically propagate the classical equations of motion for each trajectory to see if they proceed to the concerted product or fall into a stepwise intermediate basin.
- Analysis: The fraction of trajectories leading to a diradical intermediate directly quantifies the propensity for a stepwise pathway, even from a concerted-like transition structure. In the model study, 29 of 101 trajectories afforded the diradical intermediate [96].
Table 1: Key Quantitative Data from Mechanistic Competition Studies
| Reaction System | Experimental Probe | Key Quantitative Result | Interpretation | Source |
|---|---|---|---|---|
| Allenol Acetate 9 Cyclization | Primary Kinetic Isotope Effect (CH3/CD3) | kH/kD = 1.43 ± 0.12 | Highly asynchronous transition state; near concerted/stepwise boundary. | [96] |
| Model Enyne-Allene Cyclization | Direct Dynamics Trajectories | 29/101 trajectories formed diradical intermediate | Significant dynamical bifurcation from a single transition state; mechanistic mixture. | [96] |
| SN2 vs. SN2X Competition | Stereochemical Outcome & Kinetic Simulation | Halogenophilic Percentage (X%) and Relative Halogenophilicity (H) parameter developed | Quantifies intrinsic competition between simultaneous (SN2) and stepwise (SN2X) pathways. | [4] |
| SN1 Reaction Benchmark | Rate Law & Stereochemistry | Rate = k[Substrate]; Racemization (+ partial inversion) | Classic stepwise mechanism with a carbocation intermediate. | [97] [95] |
| Borderline Nucleophilic Substitution | Kinetics | Rate = k[RX] (SN1) + k'[RX][Nuc] (SN2) | Observable mixed kinetics indicating competing stepwise and concerted pathways. | [95] |
Visualization of Pathways and Workflows
Diagram 1: Competitive Landscape of Concerted vs. Stepwise Pathways
Diagram 2: Integrated Workflow for Pathway Discrimination
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagent Solutions for Competition Experiments
| Item | Function in Pathway Discrimination | Example / Note |
|---|---|---|
| Deuterated/Heavy-Isotope Labeled Substrates | Enables measurement of Kinetic Isotope Effects (KIEs) to probe bond cleavage/formation in the transition state. | CH3 vs. CD3 labeled compounds for H-transfer reactions [96]. |
| Enantiomerically Pure Chiral Electrophiles | Serves as stereochemical probes to track inversion (concerted) vs. racemization/scrambling (stepwise via intermediate). | Essential for quantifying SN2 vs. SN2X competition [4]. |
| Radical Traps (e.g., 1,4-Cyclohexadiene) | Used to intercept potential radical or diradical intermediates in stepwise mechanisms, providing evidence for their existence. | Employed in trapping studies for enyne-allene diradicals [96]. |
| Polar Aprotic & Protic Solvents | Used to modulate reaction rates and mechanism; aprotic solvents favor SN2, protic can favor SN1/stepwise paths. | Critical control variable in nucleophilic substitution studies [95]. |
| Computational Chemistry Software | For calculating transition state geometries, energies, and running quasiclassical trajectory simulations to map dynamical behavior. | Used to locate asynchronous TS and run dynamics trajectories [96]. |
| Chiral Analytical Tools (HPLC, GC) | To precisely measure enantiomeric excess or ratio in products from reactions of chiral starting materials. | Required for stereochemical analysis protocol [4]. |
| Variable-Temperature Kinetic Setup | Allows for precise measurement of rate constants and activation parameters (ΔH‡, ΔS‡), which can differ between concerted and stepwise mechanisms. | NMR spectrometers or automated sampling systems coupled to analytics. |
The study of nucleophilic substitution reactions represents a cornerstone of organic chemistry, with the stereochemical outcomes of these reactions providing the most definitive evidence for distinguishing between the two primary mechanisms: SN1 and SN2. The inversion of configuration characteristic of SN2 reactions and the racemization typical of SN1 pathways offer critical insights into the molecular events occurring during these processes. This stereochemical evidence forms the foundation for understanding reaction mechanisms and enables researchers to predict and control the outcomes of synthetic transformations. The pioneering work of Paul Walden in 1896 first demonstrated these stereochemical phenomena through the interconversion of enantiomeric malic acids, laying the experimental groundwork for our modern understanding of nucleophilic substitution mechanisms [98]. Within pharmaceutical development and synthetic chemistry, controlling stereochemistry is paramount, as the biological activity of chiral molecules often depends critically on their absolute configuration. This technical guide examines the stereochemical evidence underlying SN1 and SN2 mechanisms, providing researchers with the analytical frameworks necessary to elucidate substitution pathways in complex molecular systems.
Core Mechanistic Pathways and Stereochemical Outcomes
The SN2 Mechanism: Concerted Backside Attack
The SN2 (substitution, nucleophilic, bimolecular) mechanism proceeds through a single, concerted step in which bond formation between the nucleophile and electrophilic carbon occurs simultaneously with bond cleavage between the carbon and leaving group [99] [61]. This mechanism exhibits second-order kinetics, with the rate dependent on both the concentration of the substrate and the nucleophile [99]. The hallmark stereochemical feature of the SN2 mechanism is the complete inversion of configuration at the reaction center, often referred to as Walden inversion [3] [98].
The inversion occurs due to the backside attack mechanism, where the nucleophile approaches the carbon atom from the side opposite the departing leaving group [3]. This trajectory minimizes electronic repulsion and allows the nucleophile to donate electrons into the σ* antibonding orbital of the C-Leaving Group bond [3]. During the transition state, the carbon atom adopts a trigonal bipyramidal geometry with the nucleophile and leaving group occupying the apical positions, while the three substituents and the carbon itself begin to reorient into what will become an inverted tetrahedral geometry in the product [100].
The SN1 Mechanism: Stepwise Carbocation Formation
In contrast to the concerted SN2 pathway, the SN1 (substitution, nucleophilic, unimolecular) mechanism proceeds through a two-step process involving a discrete carbocation intermediate [99] [61]. The reaction initiates with the rate-determining dissociation of the leaving group to form a planar, sp²-hybridized carbocation intermediate, followed by nucleophilic attack on this intermediate [87]. The kinetics are first-order, dependent solely on the substrate concentration [99].
The key stereochemical consequence of this mechanism is racemization, resulting in a mixture of stereoisomeric products [99] [87]. The planar carbocation intermediate presents two geometrically equivalent faces for nucleophilic attack, with approximately equal probability of attack from either side [87]. This results in a racemic mixture (50:50 mixture of enantiomers) when substitution occurs at a chiral center, though complete racemization is often not observed in practice due to subtle stereoelectronic effects and the possibility of ion pairing that can shield one face of the carbocation [61].
Comparative Analysis: Key Stereochemical Distinctions
Table 1: Fundamental Stereochemical Differences Between SN1 and SN2 Mechanisms
| Parameter | SN2 Mechanism | SN1 Mechanism |
|---|---|---|
| Stereochemical Outcome | Complete inversion of configuration | Racemization (mixture of inversion and retention) |
| Molecularity | Bimolecular | Unimolecular (in rate-determining step) |
| Rate Law | Rate = k[Substrate][Nucleophile] | Rate = k[Substrate] |
| Reaction Steps | Single concerted step | Two-step mechanism with intermediate |
| Intermediate | None (pentacoordinate transition state only) | Carbocation |
| Geometry at Reaction Center | Trigonal bipyramidal transition state | Planar carbocation intermediate |
| Sensitivity to Steric Hindrance | High (favors unhindered substrates) | Low (relieved in carbocation formation) |
Table 2: Structural and Condition Factors Influencing Stereochemical Pathway
| Factor | Favors SN2 (Inversion) | Favors SN1 (Racemization) |
|---|---|---|
| Substrate Structure | Methyl > Primary > Secondary | Tertiary > Secondary > Primary |
| Nucleophile | Strong nucleophiles (e.g., OH⁻, CN⁻) | Weak nucleophiles (e.g., H₂O, ROH) |
| Solvent | Polar aprotic (e.g., DMSO, acetone) | Polar protic (e.g., H₂O, ROH) |
| Leaving Group | Good leaving groups required for both | Excellent leaving groups critical |
| Example Relative Rates | CH₃-X (30,000) > Et-X (1) > i-Pr-X (0.02) | t-Bu-X (1,200,000) > Et-X (1) |
Experimental Methodologies and Protocols
Kinetic Analysis of Substitution Reactions
Protocol 1: Determining Rate Law and Molecularity
- Prepare a series of reactions with varying concentrations of nucleophile while maintaining constant substrate concentration.
- Monitor reaction progress using appropriate analytical techniques (e.g., GC, HPLC, NMR) to determine initial rates.
- Plot log(rate) versus log([nucleophile]) to determine reaction order in nucleophile.
- Repeat the process varying substrate concentration with constant nucleophile concentration.
- A linear relationship with slope = 1 in both cases indicates bimolecular kinetics (SN2), while dependence only on substrate concentration indicates unimolecular kinetics (SN1) [99] [61].
Protocol 2: Stereochemical Analysis of Products
- Begin with enantiomerically pure substrate of known configuration.
- Conduct nucleophilic substitution under controlled conditions.
- Isolate and purify the substitution product.
- Determine enantiomeric composition using polarimetry, chiral HPLC, or NMR with chiral shift reagents.
- Calculate enantiomeric excess (ee): Complete inversion indicates SN2 pathway; racemization (0% ee) indicates SN1 pathway [87] [98].
Solvent Effects and Stereochemical Outcomes
Protocol 3: Probing Solvent Influence on Mechanism
- Select a secondary alkyl halide substrate (potential for both pathways).
- Perform identical substitutions in both polar protic (e.g., methanol) and polar aprotic (e.g., DMSO) solvents.
- Compare reaction rates and stereochemical outcomes.
- Polar protic solvents stabilize carbocation intermediates and favor SN1 with racemization [99].
- Polar aprotic solvents enhance nucleophilicity and favor SN2 with inversion [99] [86].
Visualization of Stereochemical Pathways
Diagram 1: Stereochemical Pathways in Nucleophilic Substitution (76 characters)
Essential Research Reagents and Materials
Table 3: Key Research Reagents for Stereochemical Studies
| Reagent/Category | Function/Application | Specific Examples |
|---|---|---|
| Chiral Substrates | Starting materials of known configuration for stereochemical tracking | (R)- or (S)-2-bromobutane, chiral tosylates |
| Nucleophiles | Varied strength to probe mechanism | Strong: CN⁻, I⁻, N₃⁻; Weak: H₂O, ROH |
| Solvent Systems | Medium to influence reaction pathway | Polar protic: H₂O, CH₃OH; Polar aprotic: DMSO, DMF, acetone |
| Leaving Groups | Critical for both mechanisms | I⁻, Br⁻, TsO⁻, MsO⁻ (tosylate, mesylate) |
| Analytical Tools | Determination of stereochemical outcome | Polarimeter, chiral HPLC columns, NMR with chiral solvating agents |
Research Context: Implications for Frontside Attack Mechanisms
The stereochemical evidence for SN2 and SN1 mechanisms provides a critical framework for investigating non-classical substitution pathways, particularly frontside attack mechanisms designated as SN2X. While traditional SN2 reactions proceed exclusively through backside attack due to electronic and steric constraints, certain structural and electronic conditions may enable frontside pathways that result in retention of configuration. Understanding the stereochemical paradigms of classical SN1 and SN2 mechanisms enables researchers to identify deviations that suggest alternative pathways.
In pharmaceutical development, where control of stereochemistry is often essential for biological activity, the ability to predict and control stereochemical outcomes of substitution reactions is paramount. The stereochemical principles governing SN1 and SN2 mechanisms provide the foundation for rational design of synthetic routes to chiral active pharmaceutical ingredients (APIs). Furthermore, the recognition of anomalous stereochemical results—such as partial retention where complete inversion is expected—can lead to the discovery of new reaction mechanisms with potential applications in asymmetric synthesis.
Stereochemical evidence remains the most definitive diagnostic tool for distinguishing between nucleophilic substitution mechanisms. The characteristic inversion of configuration in SN2 reactions and racemization in SN1 reactions provide clear mechanistic signatures that can be experimentally verified through kinetic and stereochemical analysis. As research continues into non-classical substitution pathways, including frontside attack mechanisms, these fundamental stereochemical principles provide the critical framework for identifying and characterizing new reaction pathways. For researchers in synthetic chemistry and drug development, mastery of these stereochemical concepts enables rational design of synthetic strategies and prediction of reaction outcomes in complex molecular systems.
Solvent and Subststrate Effects on Mechanism Preference
This technical guide examines the critical factors influencing mechanism preference in nucleophilic substitution reactions, with particular emphasis on the emerging frontier of frontside attack halogenophilic nucleophilic substitution (SN2X). While traditional SN2 reactions proceed via backside attack with inversion of configuration, and SN1 reactions proceed through carbocation intermediates with racemization, SN2X represents a distinct pathway involving halogenophilic attack along the C-X bond axis. This review synthesizes current understanding of how solvent properties and substrate structure dictate mechanistic pathways, providing quantitative data, experimental protocols, and visualization tools to support research in pharmaceutical development and synthetic chemistry.
Nucleophilic substitution reactions represent fundamental transformations in organic chemistry with profound implications for drug design and synthesis. The competition between SN1, SN2, and the less conventional SN2X mechanisms depends critically on substrate structure and solvent environment [70]. While SN2 reactions proceed through a concerted backside attack with second-order kinetics and inversion of configuration, SN1 reactions follow a stepwise pathway with carbocation intermediates and first-order kinetics [70] [2]. The SN2X mechanism, alternatively termed halogenophilic nucleophilic substitution, constitutes a distinct pathway wherein nucleophilic attack occurs frontside along the C-X bond direction, generating a carbanion intermediate and Nu-X electrophile that subsequently react to form products [101].
Understanding the factors governing mechanistic preference is essential for pharmaceutical scientists designing synthetic routes or investigating biochemical nucleophilic substitution reactions occurring in biological systems [2]. This review provides a comprehensive analysis of solvent and substrate effects on mechanism preference, with special attention to recent advances in SN2X reactivity and its potential applications in enantioconvergent synthesis.
Fundamental Mechanisms of Nucleophilic Substitution
SN2 Mechanism Characteristics
The SN2 (substitution nucleophilic bimolecular) mechanism proceeds through a single concerted step wherein bond formation between the nucleophile and electrophilic carbon occurs simultaneously with cleavage of the carbon-leaving group bond [102] [3]. This mechanism exhibits distinctive characteristics:
- Kinetics: Second-order rate law, Rate = k[nucleophile]×[electrophile] [70] [102]
- Stereochemistry: Backside attack results in inversion of configuration at the reaction center [1] [2]
- Steric effects: Highly sensitive to steric hindrance around the electrophilic carbon [102]
- Solvent effects: Favored by polar aprotic solvents [70] [103]
The reaction energy diagram for the SN2 pathway features a single transition state without intermediates, with the nucleophile approaching 180° relative to the leaving group, resulting in a trigonal bipyramidal transition state geometry [3] [2].
SN1 Mechanism Characteristics
The SN1 (substitution nucleophilic unimolecular) mechanism proceeds through a stepwise pathway with distinct intermediates:
- Kinetics: First-order rate law, Rate = k[electrophile] [70]
- Mechanism: Rate-determining ionization to form a carbocation intermediate, followed by nucleophilic attack [70] [2]
- Stereochemistry: Planar carbocation intermediate leads to racemization at the reaction center [70]
- Solvent effects: Favored by polar protic solvents that stabilize the ionic intermediates [70] [103]
The reaction energy diagram displays two transition states separated by a carbocation intermediate well [70].
SN2X Mechanism Characteristics
The SN2X (halogenophilic nucleophilic substitution) mechanism represents an alternative pathway with unique features:
- Reaction trajectory: Nucleophile attacks the halogen leaving group ("frontside" attack) along the C-X bond direction rather than the carbon center [101]
- Intermediates: Generates a carbanion and Nu-X electrophile that subsequently react to form product [101] [104]
- Stereochemical outcome: Distinct from both SN1 and SN2 pathways, enabling enantioconvergent transformations [101]
- Catalysis: Can be facilitated by chiral phase-transfer catalysts [101]
Computational studies reveal the importance of S···Br intermolecular halogen bonding between tertiary bromide and thiocarboxylate in facilitating efficient halogenophilic reactions [101].
Figure 1: Decision pathway for nucleophilic substitution mechanisms showing how substrate structure, solvent environment, and nucleophile characteristics direct reaction outcomes
Substrate Effects on Mechanism Preference
Substrate Structure and Steric Effects
The structure of the electrophilic substrate profoundly influences the preferred mechanism of nucleophilic substitution. The degree of substitution at the reaction center dictates the accessibility of backside attack (for SN2) versus the stability of carbocation intermediates (for SN1) [70] [102].
Table 1: Substrate Structure Effects on Nucleophilic Substitution Mechanisms
| Substrate Type | Preferred Mechanism | Relative Rate (SN2) | Key Structural Factors |
|---|---|---|---|
| Methyl | SN2 | 30 [3] | Minimal steric hindrance, no carbocation stabilization |
| Primary (1°) | SN2 | 1 [3] | Low steric hindrance, unstable carbocations |
| Secondary (2°) | SN2/E2/SN1 competition | 0.02 [3] | Moderate steric hindrance, carbocation stability balanced with backside access |
| Tertiary (3°) | SN1/E2 | ~0 [3] | High steric hindrance prevents backside attack, stable carbocations |
| Allylic/Benzylic | SN1/SN2 depending on conditions | Enhanced | Resonance-stabilized carbocations facilitate SN1, but SN2 possible |
| Neopentyl | Greatly reduced SN2 | ~10⁻⁵ [3] | Extreme steric hindrance at reaction center |
For SN2 reactions, the rate decreases dramatically with increasing substitution at the electrophilic carbon due to steric hindrance that impedes the required backside approach of the nucleophile [102] [3]. Methyl and primary substrates exhibit the fastest SN2 rates, while tertiary substrates are essentially unreactive toward the SN2 pathway [70] [3].
Substrate Effects on SN2X Reactivity
The SN2X mechanism demonstrates distinct substrate preferences compared to traditional SN2 reactions. Research has identified that brominated cyanoesters and cyanophosphonates serve as excellent tertiary electrophiles for SN2X reactions [101]. These substrates yield tertiary thioesters with high enantioselectivities when reacted under phase-transfer conditions with chiral catalysts [101].
The presence of electron-withdrawing groups adjacent to the reaction center appears to facilitate the SN2X pathway by stabilizing the carbanion intermediate generated during the halogenophilic attack [101]. This substrate preference contrasts with traditional SN2 reactions, which favor less substituted electrophiles without significant electron-withdrawing groups.
Solvent Effects on Mechanism Preference
Solvent Classification and Properties
Solvents exert profound effects on nucleophilic substitution reactions through their ability to stabilize or destabilize reactants, transition states, and intermediates. Solvents are classified into three primary categories based on polarity and hydrogen-bonding capability [70] [103]:
- Non-polar solvents (e.g., hexane, benzene, toluene): Low dielectric constants, minimal solvation of ions
- Polar protic solvents (e.g., H₂O, ROH, carboxylic acids): Contain O-H or N-H bonds, capable of hydrogen bonding, strong solvation of ions
- Polar aprotic solvents (e.g., acetone, DMSO, DMF, CH₃CN): Medium to high polarity without O-H or N-H bonds, moderate ion solvation
Table 2: Solvent Effects on Nucleophilic Substitution Mechanisms
| Solvent Type | Representative Solvents | Preferred Mechanism | Key Solvation Effects |
|---|---|---|---|
| Polar protic | H₂O, MeOH, EtOH, AcOH | SN1 | Stabilizes carbocation intermediates and leaving groups through hydrogen bonding and polar interactions |
| Polar aprotic | DMSO, DMF, acetone, CH₃CN | SN2 | Solvates cations strongly, leaving nucleophiles "naked" and highly reactive |
| Non-polar | Hexane, benzene, toluene | Neither SN1 nor SN2 favored | Poor solvation of ionic species limits both mechanisms |
| Mixed/Phase-transfer | Water-organic with phase-transfer catalysts | SN2X | Enables reactions between ionic and organic phases, facilitates halogenophilic pathway |
Molecular Basis of Solvent Effects
The influence of solvent on mechanism preference stems from differential solvation of nucleophiles and reaction intermediates:
Polar protic solvents strongly solvate anionic nucleophiles through hydrogen bonding, effectively creating a protective "cage" around the nucleophile [70] [103]. This solvation decreases nucleophilicity by increasing the effective steric bulk and reducing the electron density available for reaction. However, these solvents dramatically accelerate SN1 reactions by stabilizing the carbocation intermediate and the anionic leaving group through solvation [70].
Polar aprotic solvents solvate cations effectively through dipole interactions but cannot form strong hydrogen bonds with anions [103]. This results in "naked", highly reactive nucleophiles that dramatically enhance SN2 reaction rates [70] [103]. For example, the rate of SN2 reactions can increase by several orders of magnitude when moving from methanol to DMSO [70].
SN2X reactions benefit from phase-transfer conditions that facilitate interaction between anionic nucleophiles and organic-soluble electrophiles [101]. Chiral phase-transfer catalysts can enforce enantioselectivity in these transformations by creating a structured ionic environment that differentiates between prochiral faces [101].
Experimental Protocols and Methodologies
Kinetic Analysis of Mechanism Preference
Determining the dominant mechanism in nucleophilic substitution reactions requires careful kinetic analysis:
Protocol 1: Establishing Rate Law Dependence
- Prepare a series of reaction mixtures with constant electrophile concentration and varying nucleophile concentrations
- Monitor reaction progress using appropriate analytical techniques (GC, HPLC, NMR, or conductivity measurements)
- Determine initial rates from linear portions of concentration-time curves
- Plot initial rate versus nucleophile concentration
- Linear relationship indicates bimolecular process (SN2 or E2)
- No dependence on nucleophile concentration indicates unimolecular process (SN1 or E1)
Protocol 2: Stereochemical Analysis
- Prepare enantiomerically pure substrate (e.g., (R)- or (S)-2-bromooctane)
- Conduct nucleophilic substitution under standardized conditions
- Analyze product stereochemistry using polarimetry or chiral chromatography
- Interpret results:
- Inversion of configuration suggests SN2 pathway
- Racemization suggests SN1 pathway
- Partial inversion or unusual stereochemical outcome may indicate SN2X pathway
SN2X Reaction Under Phase-Transfer Conditions
Recent research has developed optimized conditions for enantioconvergent SN2X reactions [101]:
Reagents:
- Tertiary bromide electrophile (e.g., brominated cyanoester, 0.2 mmol)
- Thiocarboxylate nucleophile (0.24 mmol)
- Chiral phase-transfer catalyst (e.g., quaternary ammonium salt, 10 mol%)
- Biphasic solvent system: organic solvent (toluene or CH₂Cl₂) and aqueous base (NaOH or K₂CO₃ solution)
- Additives as required (e.g., crown ethers for enhanced phase transfer)
Procedure:
- Charge reaction vessel with tertiary bromide, chiral catalyst, and organic solvent
- Add aqueous phase containing base and thiocarboxylate nucleophile
- Stir vigorously at controlled temperature (0-25°C) to ensure efficient phase transfer
- Monitor reaction progress by TLC or LC-MS
- Upon completion, separate organic layer and purify products
- Analyze enantiomeric excess by chiral HPLC or NMR with chiral shift reagents
Key observations:
- Reaction proceeds with high enantioselectivity (up to 99% ee reported)
- Halogen bonding interactions crucial for transition state stabilization
- Water content and stirring efficiency critically impact reaction rate and selectivity
Solvent Effects Investigation Protocol
Systematic evaluation of solvent effects on mechanism preference:
- Select a standard substrate (e.g., 2-bromobutane for secondary halide)
- Choose nucleophile with well-characterized nucleophilicity and basicity (e.g., CN⁻ for SN2 preference, HO⁻ for E2/SN2 competition)
- Conduct parallel reactions in different solvent systems:
- Polar protic: MeOH, EtOH, i-PrOH
- Polar aprotic: DMSO, DMF, acetone, CH₃CN
- Mixed solvents: Water-acetone mixtures of varying composition
- Maintain constant temperature, concentration, and reaction time
- Quantify substitution versus elimination products using GC or NMR
- Determine relative rates and product distributions
Figure 2: Experimental workflow for determining nucleophilic substitution mechanisms, highlighting the key analytical approaches and their diagnostic outcomes
Quantitative Data and Research Tools
Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Nucleophilic Substitution Mechanisms
| Reagent Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| Solvents | DMSO, DMF, acetone, acetonitrile | Polar aprotic media for SN2 studies | Enhance nucleophile reactivity by cation solvation |
| Methanol, ethanol, water | Polar protic media for SN1 studies | Stabilize carbocation intermediates via solvation | |
| Nucleophiles | Halides (I⁻, Br⁻, Cl⁻), N₃⁻, CN⁻, RS⁻ | Poorly basic nucleophiles for SN2 studies | Minimize competing elimination pathways |
| HO⁻, RO⁻, R₂N⁻ | Strongly basic nucleophiles for E2 studies | Probe elimination versus substitution competition | |
| Electrophiles | Methyl, primary, secondary, tertiary halides | Substrate structure reactivity mapping | Establish steric and electronic effects on mechanism |
| Catalysts | Chiral quaternary ammonium salts | Phase-transfer catalysis for SN2X | Enable enantioconvergent substitutions |
| Analytical Tools | Chiral HPLC columns, polarimeter | Stereochemical analysis | Distinguish SN1 (racemization) from SN2 (inversion) |
Quantitative Reactivity Data
Recent quantitative studies of SN2X reactions have introduced parameters to characterize halogenophilicity [104]. The relative halogenophilicity (H) parameter correlates with established physical organic chemistry principles, including Hammett and Mayr postulates [104].
Studies of competing SN2 and SN2X pathways have revealed that these mechanisms exist to varying degrees in most nucleophilic substitutions and should not be considered in isolation [104]. The halogenophilic percentage (X%) can be determined through detailed kinetic simulations and stereochemical analysis of products.
For traditional SN2 reactions, quantitative relative rate data illustrates dramatic substrate effects [3]:
- Methyl bromide relative rate: ~30
- Ethyl bromide relative rate: ~1
- Isopropyl bromide relative rate: ~0.02
- Tert-butyl bromide relative rate: ~0
The preference for SN1, SN2, or SN2X mechanisms in nucleophilic substitution reactions represents a complex interplay between substrate structure, solvent environment, and nucleophile characteristics. Traditional SN2 reactions dominate with methyl and primary substrates in polar aprotic solvents, while SN1 pathways prevail with tertiary substrates in polar protic media. The recently characterized SN2X mechanism offers a distinct pathway through halogenophilic attack, enabling enantioconvergent transformations of tertiary electrophiles under phase-transfer conditions.
Understanding these mechanistic preferences provides fundamental insights for synthetic chemistry and drug development, where control over stereochemical outcomes and reaction pathways directly impacts synthetic efficiency and product profiles. Future research directions include expanding the scope of SN2X reactions, developing improved chiral catalysts for enantioconvergent processes, and applying computational methods to predict and optimize mechanism preference in complex molecular environments.
For decades, the pedagogical framework of nucleophilic substitution reactions has rested on the foundational dichotomy between the SN1 and SN2 mechanisms. The SN1 mechanism (Substitution, Nucleophilic, Unimolecular) is characterized by a stepwise process involving a rate-determining formation of a carbocation intermediate, followed by nucleophilic attack [61]. In contrast, the SN2 mechanism (Substitution, Nucleophilic, Bimolecular) proceeds via a single, concerted step featuring a backside attack that results in inversion of configuration at the stereocenter [61] [1]. This classical binary classification has successfully rationalized a vast body of chemical phenomena, including structure-reactivity relationships, kinetic data, and stereochemical outcomes. However, emerging research reveals that this two-mechanism model represents an oversimplification of a more complex and nuanced reality.
Contemporary investigations using advanced theoretical and experimental approaches have uncovered a rich spectrum of mechanistic pathways that operate between the classical SN1 and SN2 extremes. The discovery and characterization of the halogenophilic nucleophilic substitution (SN2X) reaction pathway represents a particularly significant challenge to the traditional dichotomy [5]. This mechanism, along with other nonclassical pathways, demonstrates that nucleophilic substitutions can proceed through transition states and intermediates not accounted for in conventional models. This paradigm shift has profound implications for synthetic chemistry, drug development, and our fundamental understanding of reaction mechanisms, necessitating a re-evaluation of how we conceptualize, teach, and apply nucleophilic substitution reactions in professional research settings.
Classical Mechanisms: SN1 and SN2
Fundamental Characteristics and Distinctions
The classical SN1 and SN2 mechanisms differ fundamentally in their kinetics, stereochemistry, and structural preferences. The SN1 mechanism follows first-order kinetics, where the rate of reaction depends solely on the concentration of the substrate (Rate = k [alkyl halide]) [61] [105]. This unimolecular rate law reflects the rate-determining step: the spontaneous dissociation of the leaving group to form a planar, sp²-hybridized carbocation intermediate. This carbocation can then be attacked from either face by a nucleophile, resulting in a mixture of stereochemical outcomes—typically racemization (for chiral centers) with a slight preference for inversion over retention due to ion-pair effects [61] [105].
Conversely, the SN2 mechanism exhibits second-order kinetics, with the rate dependent on both the substrate and nucleophile concentrations (Rate = k [alkyl halide][nucleophile]) [61] [105] [54]. This bimolecular process occurs in a single concerted step through a pentacoordinate transition state where bond formation with the nucleophile and bond cleavage with the leaving group occur simultaneously. The reaction proceeds exclusively via a backside attack, leading to a characteristic inversion of configuration at the reaction center, often described as an "umbrella turning inside-out" [61] [1].
Structural and Environmental Factors Governing Mechanism Selection
The propensity for a substrate to undergo SN1 versus SN2 substitution is governed by well-established structural and environmental factors, which are summarized in Table 1.
Table 1: Classical Determinants of SN1 versus SN2 Mechanism Preference
| Factor | Favors SN2 Mechanism | Favors SN1 Mechanism |
|---|---|---|
| Alkyl Halide Structure | Methyl > Primary > Secondary [61] [86] | Tertiary > Secondary > Primary [61] [86] |
| Nucleophile | High concentration of a strong nucleophile [86] [63] | Poor nucleophile (often the solvent) [86] [63] |
| Leaving Group | Good leaving groups (e.g., I⁻, Br⁻, TsO⁻) [63] | Good leaving groups (e.g., I⁻, Br⁻, TsO⁻) [63] |
| Solvent | Polar Aprotic (e.g., DMSO, DMF, acetone) [86] [63] | Polar Protic (e.g., H₂O, ROH) [86] [63] |
| Stereochemistry | Inversion of configuration [61] [1] | Racemization (or partial racemization) [61] [105] |
The underlying principles behind these trends are steric and electronic in nature. The SN2 pathway is highly sensitive to steric hindrance around the electrophilic carbon, as the nucleophile must directly approach the reaction center. Thus, less substituted alkyl halides are more reactive [61]. The SN1 pathway, however, depends critically on the stability of the carbocation intermediate; tertiary carbocations are substantially more stable than secondary or primary due to hyperconjugative stabilization and inductive effects from the alkyl groups [61] [63]. Solvent effects further modulate these preferences: polar protic solvents stabilize the ionic intermediates and transition states of the SN1 pathway through solvation, while polar aprotic solvents enhance the reactivity of anionic nucleophiles in SN2 reactions by desolvating them [86] [63].
The following diagram illustrates the fundamental stereochemical and mechanistic differences between the backside attack of the SN2 mechanism and the frontside attack that can theoretically occur in substitution reactions.
Diagram 1: Stereochemical Pathways in Nucleophilic Substitution. The diagram contrasts the concerted backside attack of the classical SN2 mechanism, leading to inversion, with a frontside attack pathway that becomes accessible in mechanisms involving cationic intermediates, leading to retention.
The SN2X Mechanism: A Third Pathway
Discovery and Defining Features
Recent quantitative studies have elucidated a distinct nucleophilic substitution pathway known as the halogenophilic nucleophilic substitution (SN2X) reaction [5]. While this pathway can yield the same final products as the classical SN2 reaction, its mechanism is fundamentally different. The SN2X pathway is characterized by an initial nucleophilic attack directed at the halogen atom (most commonly bromine or iodine) of the carbon-leaving group bond, rather than a direct backside attack on the carbon center itself [5]. This produces a metastable, hypervalent halogen intermediate, which subsequently collapses to release the product and the leaving group.
A key experimental distinction between the SN2 and SN2X pathways lies in their stereochemical behavior. The classical SN2 reaction is stereospecific, proceeding with a clean inversion of configuration due to the mandatory backside attack [1]. In contrast, the SN2X pathway, involving a pro-chiral anionic intermediate, does not necessarily lead to the same stereospecific outcome, allowing for the possibility of different stereochemical results when the two pathways compete [5]. This provides a critical experimental handle for differentiating these mechanisms and quantifying their relative contributions.
Quantitative Analysis and the Halogenophilicity Parameter
The 2024 study by Kuo et al. established a robust quantitative framework for analyzing SN2X reactions [5]. The researchers developed a procedure, supported by kinetic simulations, to measure the "halogenophilic percentage" (X%), which represents the fractional contribution of the SN2X pathway in a given reaction where both SN2 and SN2X are operative.
Furthermore, the study introduced a new intrinsic parameter termed relative halogenophilicity (H). This parameter quantifies the inherent tendency of a system to undergo the SN2X pathway and has been shown to correlate well with established physical organic chemistry principles, such as the Hammett and Mayr postulates [5]. The discovery of this parameter is significant because it provides a tool for predicting and rationalizing the behavior of substitution reactions beyond the classical models. The study concluded that SN2 and SN2X reactions exhibit similar thermodynamic and kinetic profiles, suggesting they coexist to varying degrees in many reactions and should not be considered in isolation [5].
Advanced Theoretical Insights and Borderline Mechanisms
The MEDT Perspective and Electronic Continuum
Advanced theoretical studies within the framework of Molecular Electron Density Theory (MEDT) have provided deeper insight into the electronic nature of nucleophilic substitution pathways. MEDT analyses of the transition states for both SN2 and SN2X reactions suggest they can be described as a central methyl carbocation (CH₃⁺) strongly stabilized by the simultaneous presence of the nucleophile and the leaving group through an electron density transfer process [31]. This finding challenges the traditional view of a hypervalent carbon in the SN2 transition state and reveals an electronic similarity between the molecular mechanisms of SN1 and SN2 reactions.
The MEDT perspective supports a continuum model of reactivity. The strong electronic stabilization of a tertiary carbocation, such as (CH₃)₃C⁺, allows for departure of the leaving group without significant simultaneous participation from the nucleophile, characteristic of SN1. For primary substrates, however, the departure of the leaving group is more coupled to the nucleophile's approach, leading to the SN2/SN2X-type pathways [31]. The electronic effects of substituents on the central carbon and the nature of the leaving group can effectively shift the molecular mechanism along this continuum.
Experimental Protocols for Mechanistic Discrimination
Discriminating between competing nucleophilic substitution mechanisms requires carefully designed experiments that probe kinetics, stereochemistry, and intermediate species.
1. Kinetic Isotope Effect (KIE) Measurements:
- Objective: To determine if bond formation/cleavage to a specific atom is occurring in the rate-determining step.
- Protocol: The reaction is run with a substrate containing a heavy isotope (e.g., Carbon-14, Deuterium) at a critical position (e.g., the α-carbon). The reaction rate with the isotopically labeled substrate (k) is compared to the rate with the normal substrate (k). A primary KIE (k/k > 1) indicates that the bond to the isotopically substituted atom is being broken in the rate-determining step, supporting a more SN1-like mechanism. A small or inverse KIE is often associated with concerted (SN2) mechanisms where bonds are being both formed and broken.
2. Stereochemical Analysis:
- Objective: To determine the stereochemical outcome of the substitution reaction.
- Protocol: A reaction is performed using a substrate of known enantiomeric purity and configuration. The stereochemistry of the product is analyzed using techniques such as polarimetry or chiral chromatography.
- Observation of Inversion: Supports a concerted SN2 backside attack.
- Observation of Racemization: Supports an SN1 mechanism with a planar carbocation intermediate.
- Observation of Altered/Partial Stereochemistry: Suggests a mixed or alternative mechanism, such as competition between SN2 and SN2X pathways [5].
3. Trapping of Proposed Intermediates:
- Objective: To provide direct evidence for the existence of a proposed reactive intermediate, such as a carbocation or a hypervalent halogen species.
- Protocol: A potential trapping agent (e.g., a stabilized nucleophile like sodium azide) is added to the reaction mixture in competition with the standard nucleophile. A change in product distribution or the formation of a trapped byproduct provides evidence for a discrete, diffusible intermediate (supporting SN1). The failure of a trapping agent to divert the reaction pathway suggests a concerted mechanism or an intermediate that is not freely diffusing.
The experimental workflow for a comprehensive mechanistic study integrating these techniques is visualized below.
Diagram 2: Integrated Workflow for Mechanistic Elucidation. This workflow shows the convergence of kinetic, stereochemical, trapping, and computational experiments to provide a definitive assignment of the operative substitution mechanism(s).
Research Reagents and Methodologies
The investigation of complex nucleophilic substitution mechanisms relies on a specialized toolkit of reagents, analytical techniques, and computational methods. Key resources essential for research in this field are cataloged in Table 2.
Table 2: Essential Research Reagent Solutions for Mechanistic Studies
| Reagent / Material | Function / Application in Research |
|---|---|
| Polar Aprotic Solvents (DMSO, DMF) | Solvents that enhance nucleophile reactivity by poorly solvating anions, thereby favoring SN2 and SN2X pathways. Used to probe for bimolecular mechanisms [86] [63]. |
| Polar Protic Solvents (MeOH, H₂O) | Solvents that stabilize cationic intermediates and transition states through solvation, favoring SN1 pathways. Used in solvolysis studies [86] [63]. |
| Enantiopure Alkyl Halides | Chiral substrates of defined absolute configuration (e.g., (R)- or (S)-2-bromooctane). Essential for stereochemical analysis to distinguish between inversion (SN2) and racemization (SN1) [105] [5]. |
| Stable Carbocation Salts (e.g., Trityl Tetrafluoroborate) | Salts that generate pre-formed carbocations. Used as reference compounds and in trapping experiments to model SN1 reactivity and intermediate stability. |
| Isotopically Labeled Substrates (¹⁴C, ²H) | Substrates for Kinetic Isotope Effect (KIE) studies. Allows researchers to probe whether a specific bond is being broken in the rate-determining step [31]. |
| Computational Software (Gaussian, ORCA) | Software for performing MEDT, ELF, and QTAIM analyses. Provides theoretical insights into electron density reorganization, transition state geometries, and relative energies of competing pathways [31]. |
Implications for Drug Development
The recognition of a mechanistic spectrum in nucleophilic substitution has direct and significant implications for pharmaceutical research and development. Predicting the stereochemical outcome of a substitution reaction is critical in drug synthesis, as the biological activity of a drug candidate is often highly dependent on its absolute configuration. The potential for mixed mechanisms like SN2X introduces a variable that could lead to unexpected erosion of enantiomeric purity in a synthetic step, potentially compromising the efficacy and safety profile of a drug substance [5].
Furthermore, understanding the full spectrum of substitution mechanisms enables medicinal chemists to make more informed decisions about compound design and reaction optimization. For instance, the choice of leaving group (e.g., iodide vs. bromide) or solvent (polar aprotic vs. polar protic) can not only shift the balance between SN1 and SN2 but also potentially engage the SN2X pathway, offering an alternative route that might proceed under milder conditions or with different functional group tolerance [5] [31]. This expanded mechanistic understanding provides a more sophisticated toolbox for troubleshooting synthetic routes, optimizing reaction conditions, and controlling stereochemistry in the synthesis of complex, pharmaceutically relevant molecules.
The classical SN1/SN2 dichotomy, while a valuable foundational model, is no longer sufficient to describe the full complexity of nucleophilic substitution reactions. Contemporary research, powered by quantitative kinetic studies and advanced theoretical frameworks like MEDT, has revealed a continuum of mechanistic pathways. The identification and characterization of the SN2X mechanism, along with the electronic insights provided by modern computational analyses, demonstrate that nucleophilic substitutions often proceed through a nuanced interplay of multiple pathways rather than a single, textbook mechanism.
This modern perspective demands a more sophisticated approach from researchers and industry professionals. Embracing this complexity—by employing integrated experimental protocols, leveraging quantitative parameters like relative halogenophilicity, and utilizing advanced computational tools—is essential for accurately predicting reactivity, controlling stereochemistry, and designing efficient synthetic routes. As research in this field continues to evolve, it will undoubtedly uncover further subtleties and new pathways, further enriching our understanding of one of organic chemistry's most fundamental reaction classes and driving innovation in drug discovery and materials science.
Conclusion
The strict prohibition against frontside attack in classical SN2 reactions underscores the fundamental stereoelectronic requirements of concerted nucleophilic substitution, where backside displacement remains the exclusive pathway due to optimal orbital overlap and minimized steric and electronic repulsion. This mechanistic understanding provides critical insights for drug development professionals who require precise stereochemical control in pharmaceutical synthesis. Future research directions should explore borderline systems where structural modifications or medium effects might enable unusual reaction pathways, investigation of substitution mechanisms at non-carbon centers with potential biomedical applications, development of computational methods for predicting stereochemical outcomes in complex molecular environments, and design of engineered reaction systems that exploit these fundamental principles for innovative synthetic methodologies. The continued elucidation of these fundamental reaction mechanisms remains essential for advancing synthetic strategy in medicinal chemistry and drug development.
References
- 1. Stereochemistry \(S_N2\) Reactions [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Reactions/Substitution_Reactions/SN2/Sterochemistry]
- 2. 8.2: Two Mechanistic Models for Nucleophilic Substitution [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_v2.0_(Soderberg)/08%3A_Nucleophilic_Substitution_Reactions/8.02%3A_Two_Mechanistic_Models_for_Nucleophilic_Substitution]
- 3. The SN2 Reaction Mechanism [https://www.masterorganicchemistry.com/2012/07/04/the-sn2-mechanism/]
- 4. A Quantitative Study of the Halogenophilic Nucleophilic ... [https://pubmed.ncbi.nlm.nih.gov/39652785/]
- 5. A Quantitative Study of the Halogenophilic Nucleophilic ... [https://oar.a-star.edu.sg/communities-collections/articles/21424?collectionId=32]
- 6. 8.5: Nucleophilic Substitution - 2nd Order [https://chem.libretexts.org/Courses/Providence_College/Organic_Chemistry_I/08%3A_Substitution_Reactions/8.05%3A_Nucleophilic_Substitution_-_2nd_Order]
- 7. SN1 reaction [https://en.wikipedia.org/wiki/SN1_reaction]
- 8. Gas phase SN of halide ions with trifluoromethyl halides... 2 reactions [https://pubmed.ncbi.nlm.nih.gov/16435795/]
- 9. Nucleophilic Substitution (SN2) - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC6001448/]
- 10. Numerical separation of the front-side attack and double- ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261420306953]
- 11. Stereoelectronic Effect - an overview [https://www.sciencedirect.com/topics/chemistry/stereoelectronic-effect]
- 12. - Wikipedia Walden inversion [https://en.wikipedia.org/wiki/Walden_inversion]
- 13. : Walden , Examples & Key Concepts Explained Inversion Mechanism [https://www.vedantu.com/chemistry/walden-inversion]
- 14. A Quantitative Study of the Halogenophilic Nucleophilic ... [https://www.nature.com/nature-index/article/10.1021/jacs.4c12463]
- 15. SN2 reaction [https://en.wikipedia.org/wiki/SN2_reaction]
- 16. Capturing penta-coordinate carbon! (Part 2). - Henry Rzepa's ... [https://www.ch.ic.ac.uk/rzepa/blog/?p=811]
- 17. Pentacoordinate Carbon? - Computational Organic Chemistry [https://comporgchem.com/blog/archives/385]
- 18. Capturing penta-coordinate carbon! (Part 1). [https://www.ch.ic.ac.uk/rzepa/blog/?p=783]
- 19. Pentacoordinate Carbon - (Organic Chemistry) [https://fiveable.me/key-terms/organic-chem/pentacoordinate-carbon]
- 20. Transition States in Chemical Reactions [https://people.chem.ucsb.edu/kahn/kalju/chem126/public/qm_ts_optim.html]
- 21. A pentacoordinate carbon - Computational Organic Chemistry [https://comporgchem.com/blog/archives/3213]
- 22. Discovery of a New Class of Organic Reactions [https://tanchoonhong.wixsite.com/tchlab/post/discovery-of-a-new-class-of-organic-reactions]
- 23. 6.1: The SN2 Reaction [https://chem.libretexts.org/Courses/University_of_Alberta_Augustana_Campus/AUCHE_250%3A_Organic_Chemistry_I/06%3A_Substitutions/6.01%3A_The_SN2_Reaction]
- 24. 11.2: The SN2 Reaction [https://chem.libretexts.org/Courses/Athabasca_University/Chemistry_350%3A_Organic_Chemistry_I/11%3A_Reactions_of_Alkyl_Halides-_Nucleophilic_Substitutions_and_Eliminations/11.02%3A_The_SN2_Reaction]
- 25. 11.3: Characteristics of the SN2 Reaction [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(OpenStax)/11%3A_Reactions_of_Alkyl_Halides-_Nucleophilic_Substitutions_and_Eliminations/11.03%3A_Characteristics_of_the_SN2_Reaction]
- 26. Stereospecific nucleophilic substitution at tertiary and ... [https://pubs.rsc.org/en/content/articlelanding/2020/sc/d0sc02562c]
- 27. Stereospecific nucleophilic substitution at quaternary ... [https://www.sciencedirect.com/science/article/pii/S2451929423000219]
- 28. SN2 versus SN2′ Competition - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9295157/]
- 29. Contact ion-pair SN2 reactions activated by Lewis Base ... [https://www.nature.com/articles/s41467-024-55795-6]
- 30. 7.2: SN2 Reaction Mechanism, Energy Diagram and ... [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_I_(Liu)/07%3A_Nucleophilic_Substitution_Reactions/7.02%3A_SN2_Reaction_Mechanism_Energy_Diagram_and_Stereochemistry]
- 31. Advanced Molecular Electron Density Theory Study of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12290940/]
- 32. Refining potential energy surface through dynamical ... [https://www.nature.com/articles/s41467-025-56061-z]
- 33. Determination of a Potential Energy Surface for CO2Using ... [https://www.sciencedirect.com/science/article/abs/pii/S0022285299978165]
- 34. 5: Chemical Kinetics, Reaction Mechanisms, and ... [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Thermodynamics_and_Chemical_Equilibrium_(Ellgen)/05%3A_Chemical_Kinetics_Reaction_Mechanisms_and_Chemical_Equilibrium]
- 35. Reaction progress kinetic analysis [https://en.wikipedia.org/wiki/Reaction_progress_kinetic_analysis]
- 36. An Asymmetric Chemical Reaction With Intriguing ... [https://www.technologynetworks.com/drug-discovery/news/an-asymmetric-chemical-reaction-with-intriguing-reaction-pathways-315293]
- 37. The stereochemistry of substitution at S(VI) [https://pubs.rsc.org/en/content/articlehtml/2025/qo/d5qo01043h]
- 38. The stereochemistry of substitution at S(vi) [https://pubs.rsc.org/en/content/articlelanding/2025/qo/d5qo01043h]
- 39. Enantioconvergent nucleophilic substitution via synergistic ... [https://www.nature.com/articles/s41929-024-01288-0]
- 40. Fifty years of nucleophilic substitution in the gas phase - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9291629/]
- 41. New model predicts a chemical reaction's point of no return [https://news.mit.edu/2025/new-model-predicts-chemical-reactions-no-return-point-0423]
- 42. Guessing Transition States - Rowan Scientific [https://rowansci.com/blog/guessing-transition-states]
- 43. Double-Ended TS Search and the Invisible Work of Computer ... [https://rowansci.substack.com/p/double-ended-ts-search-and-the-invisible]
- 44. Application of neural network potentials to modelling ... [https://pubs.rsc.org/en/content/articlelanding/2025/cc/d5cc02090e]
- 45. Real-time capture of reactive intermediates in an enzymatic reaction: insights into a P450-catalyzed oxidation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12108963/]
- 46. Identifying reactive intermediates by mass spectrometry - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8162775/]
- 47. Isotope Labeling Techniques: A Comprehensive Guide ... [https://www.metwarebio.com/isotope-labeling-guide-applications/]
- 48. Isotopic labelings for mechanistic studies [https://pubmed.ncbi.nlm.nih.gov/38942502/]
- 49. Principles and Characteristics of Isotope Labeling [https://www.creative-proteomics.com/resource/principles-characteristics-isotope-labeling.htm]
- 50. Isotope dilution mass spectrometry for absolute ... [https://www.sciencedirect.com/science/article/abs/pii/S1874391913005605]
- 51. Stable-isotope dilution LC–MS for quantitative biomarker ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2843934/]
- 52. Impact of kinetic isotope effects in isotopic studies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4583766/]
- 53. Stable Isotope Labeling Strategies [https://proteomicsresource.washington.edu/protocols03/isotopic_labeling.php]
- 54. A Closer Look at the Nucleophilic Substitution Mechanism ... [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Vollhardt_and_Schore)/06._Properties_and_Reactions_of_Haloalkanes%3A_Bimolecular_Nucleophilic_Substitution/6-04_A_Closer__Look__at_the__Nucleophilic_Substitution_Mechanism%3A_Kinetics]
- 55. The isocyanide SN2 reaction [https://www.nature.com/articles/s41467-023-41253-2]
- 56. The isocyanide SN2 reaction [https://pubmed.ncbi.nlm.nih.gov/37726293/]
- 57. [PDF] The isocyanide SN2 reaction [https://www.semanticscholar.org/paper/115e2be12ab0cc26ea00742cae958a7701848e23]
- 58. The isocyanide SN2 reaction [https://research.rug.nl/en/publications/the-isocyanide-sn2-reaction/]
- 59. SN2 Mechanism and Kinetics [https://learn.openochem.org/learn/first-semester-topics/substitutions-and-eliminations/sn2-reaction/sn2-mechanism-and-kinetics]
- 60. 11.2: The SN2 Reaction [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/11%3A_Reactions_of_Alkyl_Halides-_Nucleophilic_Substitutions_and_Eliminations/11.02%3A_The_SN2_Reaction]
- 61. Comparing The SN1 vs Sn2 Reactions [https://www.masterorganicchemistry.com/2012/08/08/comparing-the-sn1-and-sn2-reactions/]
- 62. 10.4: Effect of sterics on Sn2 reactions [https://chem.libretexts.org/Courses/Purdue/Purdue%3A_Chem_26605%3A_Organic_Chemistry_II_(Lipton)/Chapter_10._Nucleophilic_Substitution/10.4%3A_Effect_of_sterics_on_Sn2_reactions]
- 63. Nucleophilic Substitution (SN1, SN2) [https://www.organic-chemistry.org/namedreactions/nucleophilic-substitution-sn1-sn2.shtm]
- 64. Up-ending a Fundamental Reaction in Organic Chemistry [https://blogs.ntu.edu.sg/science/2019/01/30/189/]
- 65. What Makes A Good Nucleophile? [https://www.masterorganicchemistry.com/2012/06/18/what-makes-a-good-nucleophile/]
- 66. Nucleophile [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Reactions/Substitution_Reactions/SN2/Nucleophile]
- 67. Ch 8: Nucleophiles [https://www.chem.ucalgary.ca/courses/350/Carey5th/Ch08/ch8-5.html]
- 68. 11.3: Characteristics of the SN2 Reaction [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/11%3A_Reactions_of_Alkyl_Halides-_Nucleophilic_Substitutions_and_Eliminations/11.03%3A_Characteristics_of_the_SN2_Reaction]
- 69. 11.3: Characteristics of the SN2 Reaction [https://chem.libretexts.org/Courses/Athabasca_University/Chemistry_350%3A_Organic_Chemistry_I/11%3A_Reactions_of_Alkyl_Halides-_Nucleophilic_Substitutions_and_Eliminations/11.03%3A_Characteristics_of_the_SN2_Reaction]
- 70. 7.5: SN1 vs SN2 [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_I_(Liu)/07%3A_Nucleophilic_Substitution_Reactions/7.05%3A_SN1_vs_SN2]
- 71. Exploring borderline SN1–SN2 mechanisms: the role of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10841197/]
- 72. Polar Protic? Polar Aprotic? Nonpolar? All About Solvents [https://www.masterorganicchemistry.com/2012/04/27/polar-protic-polar-aprotic-nonpolar-all-about-solvents/]
- 73. Polar Protic and Polar Aprotic Solvents [https://www.chemistrysteps.com/polar-protic-and-polar-aprotic-solvents/]
- 74. Sn2 Reaction Polar Aprotic Solvents [https://rt-students.com/sn2-reaction-polar-aprotic-solvents]
- 75. SN2 Effect of Solvent [https://learn.openochem.org/learn/first-semester-topics/substitutions-and-eliminations/sn2-reaction/sn2-effect-of-solvent]
- 76. The Sn2 Reaction: A Theoretical-Computational Analysis ... [https://www.mdpi.com/2504-3900/41/1/81]
- 77. Leaving group [https://en.wikipedia.org/wiki/Leaving_group]
- 78. Leaving Groups - Chemistry LibreTexts [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Reactions/Substitution_Reactions/SN2/Leaving_Groups]
- 79. What Makes A Good Leaving Group [https://www.masterorganicchemistry.com/2011/04/12/what-makes-a-good-leaving-group/]
- 80. Two Types of Nucleophilic Substitution Reactions [https://www.masterorganicchemistry.com/2012/06/27/two-types-of-substitution-reactions/]
- 81. 7.3: Other Factors that Affect SN2 Reactions [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_I_(Liu)/07%3A_Nucleophilic_Substitution_Reactions/7.03%3A_Other_Factors_that_Affect_SN2_Reactions]
- 82. Nucleophilic Substitution at Heteroatoms—Identity ... [https://www.mdpi.com/1420-3049/27/3/599]
- 83. Nucleophilic Substitution Reaction: Definition, Types, SN1 ... [https://www.aakash.ac.in/important-concepts/chemistry/nucleophilic-substitution-reaction]
- 84. 4.5: Factors affecting the SN2 Reaction [https://chem.libretexts.org/Courses/Brevard_College/CHE_202%3A_Organic_Chemistry_II/04%3A_Substitution_and_Elimination_reactions/4.05%3A_Factors_affecting_the_SN2_Reaction]
- 85. 8.2. Physical chemistry for SN2 and SN1 reactions [https://courses.lumenlearning.com/suny-potsdam-organicchemistry/chapter/8-2-physical-chemistry-for-sn2-and-sn1-reactions/]
- 86. 7.12: Comparison of SN1 and SN2 Reactions [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Wade)_Complete_and_Semesters_I_and_II/Map%3A_Organic_Chemistry_(Wade)/07%3A_Alkyl_Halides-_Nucleophilic_Substitution_and_Elimination/7.12%3A_Comparison_of_SN1_and_SN2_Reactions]
- 87. 7.3: Stereochemical Consequences of SN1 Reactions [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Vollhardt_and_Schore)/07._Further_Reactions_of_Haloalkanes%3A_Unimolecular_Substitution_and_Pathways_of_Elimination/7.3%3A_Stereochemical_Consequences_of_SN1__Reactions]
- 88. Understanding SN1 vs. SN2 Reactions [https://chemistrytalk.org/sn1-vs-sn2/]
- 89. Thermodynamic Insights into Protein Dynamics and Drug ... [https://esmed.org/MRA/mra/article/view/6867]
- 90. 11.4: The SN1 Reaction [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/11%3A_Reactions_of_Alkyl_Halides-_Nucleophilic_Substitutions_and_Eliminations/11.04%3A_The_SN1_Reaction]
- 91. 5.7: Reactive Intermediates - Carbocations [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Wade)_Complete_and_Semesters_I_and_II/Map%3A_Organic_Chemistry_(Wade)/05%3A_An_Introduction_to_Organic_Reactions_using_Free_Radical_Halogenation_of_Alkanes/5.07%3A_Reactive_Intermediates_-_Carbocations]
- 92. The Carbocation Intermediate In The SN1, E1, and Alkene ... [https://www.masterorganicchemistry.com/2015/04/21/carbocations-and-the-sn1-e1-and-alkene-addition-reactions/]
- 93. making sense of multiple transition states in parallel and series [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d4cs00868e]
- 94. New deep learning method identifies transition states in ... [https://chem.wisc.edu/2025/11/05/research-new-deep-learning-method-identifies-transition-states-in-protein-conformational-changes/]
- 95. Nucleophilic substitution [https://en.wikipedia.org/wiki/Nucleophilic_substitution]
- 96. 'Concerted' Transition State, Stepwise Mechanism. ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2501108/]
- 97. The SN1 Reaction Mechanism [https://www.masterorganicchemistry.com/2012/07/13/the-sn1-mechanism/]
- 98. 11.1 The Discovery of Nucleophilic Substitution Reactions [https://openstax.org/books/organic-chemistry/pages/11-1-the-discovery-of-nucleophilic-substitution-reactions]
- 99. Difference Between SN and 1 Reactions: Key Characteristics... SN 2 [https://www.vedantu.com/jee-advanced/chemistry-difference-between-sn1-and-sn2]
- 100. Perez-Benitez, model for the stereochemistry of the SN and... 1 [http://www.edu.utsunomiya-u.ac.jp/chem/v12n1/Perez_Benitez8017/Perez8017.html]
- 101. Enantioconvergent halogenophilic nucleophilic substitution ... [https://dr.ntu.edu.sg/entities/publication/4d5d8d10-5e5c-4472-ae77-f9b7caaada7b]
- 102. 6.10: Structure and SN2 Reactivity: The Substrate [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Vollhardt_and_Schore)/06._Properties_and_Reactions_of_Haloalkanes%3A_Bimolecular_Nucleophilic_Substitution/6.10%3A_Structure_and_SN2_Reactivity%3A_The_Substrate]
- 103. The Role of Solvent in SN1, SN2, E1 and E2 Reactions [https://www.chemistrysteps.com/sn1-sn2-e1-e2-effect-of-solvent/]
- 104. A quantitative study of the halogenophilic nucleophilic ... [https://dr.ntu.edu.sg/entities/publication/cb6f7023-716f-4eb2-bc48-be69b7e0f632]
- 105. SN1 and SN2 reaction – Kinetics, Mechanism ... [https://chemicalnote.com/sn1-and-sn2-reaction-kinetics-mechanism-stereochemistry-and-reactivity/]