Reaction Mechanisms in Organic Chemistry: From Fundamental Concepts to Drug Discovery Applications
This article provides a comprehensive overview of organic reaction mechanisms, tailored for researchers, scientists, and drug development professionals.
Reaction Mechanisms in Organic Chemistry: From Fundamental Concepts to Drug Discovery Applications
Abstract
This article provides a comprehensive overview of organic reaction mechanisms, tailored for researchers, scientists, and drug development professionals. It bridges fundamental concepts with cutting-edge applications in medicinal chemistry, covering the step-by-step processes of key reactions, modern methodological approaches for mechanism elucidation, and strategies for optimizing challenging transformations. A significant focus is placed on the critical role of mechanistic understanding in drug design, featuring case studies of clinical candidates and exploring the application of mechanisms in new therapeutic modalities like PROTACs and DNA-encoded libraries. The content also addresses the importance of mechanism validation and comparative analysis to ensure predictive accuracy in complex biological systems, offering a holistic resource for advancing drug discovery efforts.
Core Principles and Key Reactive Intermediates in Organic Mechanisms
Understanding reaction mechanisms is fundamental to advancing research in organic chemistry, particularly in fields like drug development where predicting and controlling molecular transformations is paramount. A reaction mechanism is a sequential account of the elementary steps that describe the pathway from reactants to products at the atomic and molecular level [1]. This detailed mapping is crucial for researchers and scientists, as it provides a predictive framework for designing novel synthetic routes, optimizing reaction conditions, and developing new therapeutic agents. At its core, every organic reaction involves the reorganization of atoms, a process governed by the breaking of existing bonds, the formation of new ones, and the movement of electrons that facilitates this molecular rearrangement [2]. This whitepaper delineates these core principles, providing an in-depth technical guide framed within the context of modern organic chemistry research.
Fundamental Components of a Reaction Mechanism
Bond-Breaking and Bond-Making Processes
The transformation of reactants into products is characterized by changes in molecular connectivity. These changes are categorized based on how electron pairs are distributed during bond cleavage and formation.
- Homolytic Bond Cleavage: In this process, a covalent bond breaks symmetrically, with each atom retaining one electron from the bonding pair. This generates highly reactive, neutral intermediates called radicals. The process is often represented as: A:B → A• + B•. Homolytic cleavage is typically induced by heat, light (photolysis), or radical initiators and is a key step in radical chain reactions relevant to polymer science and certain biochemical pathways [2].
- Heterolytic Bond Cleavage: This process involves an unsymmetrical bond break, where the electron pair is completely taken by one of the two atoms. This results in the formation of a charged species: a cation (if the atom that did not take the electrons was more electronegative) and an anion (if the atom that took the electrons was more electronegative), represented as: A:B → A⁺ + :B⁻. Heterolytic cleavage is prevalent in polar reactions, which are commonplace in solutions and biological systems [2] [3].
- Bond Formation: The formation of a new bond occurs when an electron-rich site (a nucleophile, possessing a lone pair or π-bond) donates electrons to an electron-deficient site (an electrophile). This can be the coupling of two radicals or the interaction between a nucleophile and an electrophile, the latter being a cornerstone of ionic reaction mechanisms [3].
The Language of Electron Movement
Electron-pushing arrows are the universal "bookkeeping" device used by chemists to track the flow of electrons during these processes, providing tremendous value for both understanding and predicting chemical reactivity [3].
- The Curved Arrow: A curved arrow depicts the movement of an electron pair from a source (a lone pair or a bond) to a destination (an atom or a new bond location).
- Fundamental Arrow Patterns:
- Bond Breaking (Heterolytic): An arrow originates from the center of a bond (signifying the electron pair) and points to a specific atom. This shows one atom taking the bond's electron pair, resulting in heterolytic cleavage [3].
- Bond Making: An arrow originates from a lone pair on an atom (or sometimes a π-bond) and points to the space between two atoms to signify the formation of a new bond [3].
- Bond Movement (Synchronous Processes): This is a shorthand notation that represents simultaneous bond breaking and bond making in a concerted step, such as in a substitution (S_N2) reaction [3].
Table 1: Taxonomy of Electron-Pushing Arrows in Mechanism Depiction
| Arrow Type | Origin | Destination | Represented Process | Formal Charge Change |
|---|---|---|---|---|
| Bond Breaking | Center of a bond | A specific atom | Heterolytic cleavage | Starting atom: +1; Terminal atom: -1 |
| Bond Making | Lone pair on an atom | Space between two atoms | Formation of a new covalent bond | Starting atom: +1; Terminal atom: -1 |
| Bond Movement | Center of a bond | A sextet atom | Concerted bond break/formation | No change on central atoms; changes on terminal atoms |
Advanced Representation: The Imaginary Transition Structure (ITS)
Moving beyond traditional arrow notation, the Imaginary Transition Structure (ITS) model, pioneered by Shinsaku Fujita, offers a powerful graph-theoretical approach to representing reactions. An ITS encodes the entire reaction—reactants, products, and the atom mappings between them—within a single, unified, undirected graph [4].
ITS Bond Classification and Data Model
In an ITS graph, edges ("ITS bonds") are classified based on their role in the reaction and are labeled with an ordered pair of integers (a, b) [4]:
- a: Denotes the bond order in the starting material.
- b: Denotes the change in bond order caused by the reaction. The bond order in the product is simply the sum (a + b).
Table 2: ITS Bond Labeling and Classification System
| Bond Type | ITS Label (a, b) | Starting Order | Product Order | Visual Representation |
|---|---|---|---|---|
| Par-bond | (1, 0), (2, 0), (3, 0) | a | a (Unchanged) | Solid Line |
| In-bond | (0, +1), (0, +2) | 0 | b (Formed) | Dashed Line |
| Out-bond | (1, -1), (2, -1), (2, -2) | a | a + b (Broken) | Dotted Line |
Reaction Center (RC) Graphs and Classification
A critical subgraph of the ITS is the Reaction Center (RC) Graph, which contains only the dynamic nodes and edges—those atoms and bonds directly involved in the electron redistribution [4]. The RC graph is the reaction analog of a molecular functional group. Fujita classified reactions based on "stringity," which refers to the number of alternating sequences (strings) of in-bonds and out-bonds within the RC graph. A key graph-theoretical insight is that every valid RC graph must have a perfect matching between its in-bonds and out-bonds, a concept familiar to organic chemists as the principle of "pushing electrons" [4].
Diagram 1: Generalized Workflow of a Multi-Step Reaction Mechanism
Experimental and Computational Methodologies
Elucidating a reaction mechanism requires a combination of experimental kinetic studies, isotopic labeling, and advanced computational modeling.
Kinetic Analysis and Isotopic Labeling
Traditional methods involve measuring reaction rates under varying concentrations of reactants to determine the order of the reaction, which provides clues about the rate-determining step and the molecularity of the reaction [5]. Isotopic labeling, particularly with deuterium (²H) or heavy oxygen (¹⁸O), allows researchers to track the fate of specific atoms during a reaction. For instance, kinetic isotope effects (KIEs) compare the reaction rates of labeled versus unlabeled substrates; a significant KIE suggests the bond to the labeled atom is being broken in the rate-determining step [6].
Computational Studies using Density Functional Theory (DFT)
Modern mechanistic research heavily relies on computational chemistry. Density Functional Theory (DFT) is a primary tool for modeling reaction pathways [5] [1]. Researchers use DFT to calculate the energies and geometries of reactants, transition states, intermediates, and products along a proposed reaction coordinate. This allows for the visualization of bond lengths and atomic charges at each stage and provides a quantitative measure of the energy barrier (activation energy) for each step [5].
Table 3: Key Reagent Solutions for Mechanistic Studies
| Research Reagent / Material | Function in Mechanistic Analysis | Example Application |
|---|---|---|
| Deuterated Solvents (e.g., CDCl₃) | Solvent for NMR spectroscopy; allows tracking of H/D exchange. | Probing reaction intermediates via in-situ NMR. |
| Isotopically Labeled Substrates | Acts as a "tracer" for specific atoms within a reaction pathway. | Determining Kinetic Isotope Effects (KIEs) to identify bond-breaking in the rate-determining step. |
| Density Functional Theory (DFT) Software | Computational modeling of energy surfaces and transition states. | Calculating activation energies and optimizing geometries of proposed intermediates [5]. |
| Lewis Acid Catalysts (e.g., BF₃, AlCl₃) | Activates substrates by binding to nucleophilic sites (lone pairs, π-bonds). | Initiating cationic cascades (e.g., Friedel-Crafts alkylation) for complexity building [3]. |
| Stoichiometric Organometallics (e.g., n-BuLi) | Strong base or nucleophile; can initiate carbanion-based mechanisms. | Deprotonation to generate reactive enolates or for metal-halogen exchange [3]. |
Diagram 2: Mechanistic Elucidation via Integrated Experimental and Computational Workflow
Current Research and Application in Drug Development
The principles of mechanism are actively applied in cutting-edge research to solve complex challenges in synthesis and catalysis.
Case Study: Ammonia Decomposition for Hydrogen Carrier Systems
In the context of clean energy, which has implications for pharmaceutical manufacturing processes, ammonia decomposition has emerged as an important route for hydrogen production. Mechanistic studies using DFT on non-precious metal catalysts like Fe, Co, and Ni have revealed that the reaction proceeds via a stepwise dehydrogenation mechanism [5]. The initial step involves the adsorption and dehydrogenation of NH₃ to NH₂* and H, followed by further dehydrogenation to NH and finally the recombination of N* atoms to form N₂. Understanding this mechanism allows for the rational design of more efficient and cheaper catalysts, which is critical for large-scale application [5].
Emerging Trends: CGRs and Machine Learning
The ITS concept has been revitalized in the form of the Condensed Graph of Reaction (CGR), which is used in machine learning (ML) applications [4]. In this approach, reactions are represented in a SMILES-like notation that encodes dynamic bonds (e.g., [->] for a broken bond, [>-] for a formed bond). These CGRs can be fed into ML models, such as autoencoders, to explore chemical reaction space and predict novel reactions not present in the training data, offering a powerful tool for de novo reaction design in medicinal chemistry [4].
A rigorous and detailed understanding of reaction mechanisms—encompassing the intricacies of bond-breaking, bond-forming, and electron movement—is indispensable for progress in organic chemistry and drug development. From the foundational use of electron-pushing arrows to the sophisticated graph-based models of ITS and CGRs, the tools for describing and predicting reactivity continue to evolve. As computational power and machine learning algorithms advance, the integration of these mechanistic principles with data-driven approaches will undoubtedly accelerate the discovery and optimization of new chemical transformations, thereby fueling innovation in the synthesis of complex molecules and therapeutic agents.
Within organic chemistry research and pharmaceutical development, nucleophilic substitution reactions represent a cornerstone methodology for constructing complex molecular architectures. The strategic choice between the unimolecular (SN1) and bimolecular (SN2) pathways is not merely an academic exercise but a critical decision point in synthetic design that directly impacts yield, stereochemical outcome, and scalability. This guide provides an in-depth analysis of these fundamental mechanisms, empowering researchers to make informed predictions and optimize reaction conditions for advanced synthetic applications, including the synthesis of active pharmaceutical ingredients (APIs) and novel chemical entities.
The intellectual framework for understanding these reactions rests on the principle that mechanism dictates outcome. By systematically evaluating substrate structure, nucleophile strength, solvent environment, and leaving group ability, scientists can navigate the complex energy landscapes that differentiate these pathways, enabling precise control over molecular structure in drug discovery and development campaigns.
Core Mechanisms and Kinetic Profiles
SN1 Mechanism: A Stepwise Pathway
The SN1 (Substitution Nucleophilic Unimolecular) mechanism proceeds through a two-step, dissociative pathway where rate-determining ionization forms a planar carbocation intermediate prior to nucleophilic attack [7] [8].
Mechanistic Steps:
- Ionization (Rate-Determining): Slow, spontaneous dissociation of the carbon-leaving group (C–LG) bond yields a carbocation intermediate and the departed leaving group. This step has high activation energy due to the instability of the developing carbocation [7].
- Nucleophilic Attack: Fast addition of a nucleophile to the electrophilic carbocation. The nucleophile can approach with equal probability from either face of the trigonal planar carbocation, leading to potential loss of stereochemical integrity [7] [8].
Kinetics: The SN1 reaction follows first-order kinetics, where the rate depends solely on the concentration of the substrate alkyl halide [7] [9] [10]. The rate law is expressed as: Rate = k [Alkyl Halide] This unimolecular rate law reflects the rate-determining step's dependence on the substrate's spontaneous ionization [7].
SN2 Mechanism: A Concerted Pathway
The SN2 (Substitution Nucleophilic Bimolecular) mechanism occurs via a single, concerted step featuring a nucleophilic backside attack that coincides with leaving group departure [7] [8].
Mechanistic Steps:
- Concerted Displacement: A single transition state characterizes the synchronous process where the nucleophile attacks the electrophilic carbon from the side opposite the leaving group (backside attack), forming a partial bond (C–Nu), while the leaving group departs with simultaneous breaking of the C–LG bond. The carbon center adopts a trigonal bipyramidal geometry in the transition state [7].
- Inversion of Configuration: The reaction proceeds with complete inversion of stereochemistry at the reaction center, analogous to an umbrella turning inside out in the wind [7].
Kinetics: The SN2 reaction follows second-order kinetics, with the rate proportional to the concentrations of both the substrate and the nucleophile [7] [8] [10]. The rate law is expressed as: Rate = k [Alkyl Halide] [Nucleophile] This bimolecular rate law indicates that both reactants are involved in the rate-determining step [7].
Comparative Analysis: Determinants of Reaction Pathway
The divergence between SN1 and SN2 pathways is governed by distinct structural and experimental parameters. The following comparative table synthesizes the core differentiating factors critical for reaction prediction and design.
Table 1: Comprehensive Comparison of SN1 and SN2 Reaction Parameters
| Parameter | SN1 Mechanism | SN2 Mechanism |
|---|---|---|
| Molecularity | Unimolecular [7] [10] | Bimolecular [7] [10] |
| Rate Law | Rate = k [Alkyl Halide] [7] [9] | Rate = k [Alkyl Halide][Nucleophile] [7] [8] |
| Reaction Steps | Two-step (stepwise) with intermediate [7] [10] | One-step (concerted) [7] [10] |
| Key Intermediate | Carbocation [7] [8] | None (Pentacoordinate Transition State) [7] |
| Substrate Reactivity | Tertiary > Secondary > Primary [7] [8] | Methyl > Primary > Secondary >> Tertiary [7] [11] |
| Stereochemistry | Racemization (or partial inversion) [7] [8] | Inversion of configuration [7] [8] |
| Nucleophile | Weak nucleophile acceptable (often the solvent) [9] [10] | Strong nucleophile required [9] [10] |
| Solvent Preference | Polar Protic (e.g., H₂O, ROH) [9] [10] | Polar Aprotic (e.g., DMSO, DMF, acetone) [9] [10] |
| Leaving Group | Critical; excellent leaving group required [7] [9] | Critical; excellent leaving group required [7] [9] |
Substrate Structure: A Primary Determinant
The structure of the alkyl halide substrate is arguably the most significant factor in determining the operative mechanism, primarily due to its profound effect on carbocation stability (for SN1) and steric accessibility (for SN2).
Table 2: Structural Effects on Substitution Pathway
| Substrate Type | SN1 Reactivity | Rationale | SN2 Reactivity | Rationale |
|---|---|---|---|---|
| Methyl | Not reactive [10] | Methyl carbocation is highly unstable [11] | Very fast [10] | Minimal steric hindrance to backside attack [7] |
| Primary (1°) | Slow [10] | Primary carbocations are unstable [11] | Fast [10] | Low steric hindrance around the carbon center [7] |
| Secondary (2°) | Moderate [10] | Stabilized by hyperconjugation and solvent effects [7] | Moderate [10] | Steric hindrance becomes significant but not prohibitive [7] |
| Tertiary (3°) | Fast [10] | Tertiary carbocations are highly stabilized by hyperconjugation [7] [11] | Very slow or not at all [10] | Severe steric hindrance blocks the required backside attack [11] |
Solvent Effects and Nucleophile Strength
The reaction medium and nucleophile identity provide powerful levers for directing reaction pathways.
Solvent Effects: Polar protic solvents (e.g., water, ethanol) solvate and stabilize the ionic intermediates and transition states in SN1 reactions, dramatically accelerating the rate of carbocation formation [9] [10]. Conversely, these solvents strongly solvate nucleophiles via hydrogen bonding, reducing their reactivity and disfavoring SN2 pathways. Polar aprotic solvents (e.g., dimethyl sulfoxide (DMSO), dimethylformamide (DMF), acetone) do not solvate nucleophiles as effectively, leaving them "naked" and more reactive, thereby favoring the SN2 mechanism [9] [10].
Nucleophile Strength: SN2 reactions require strong nucleophiles (e.g., HO⁻, CN⁻, I⁻, RO⁻) to facilitate the concerted displacement [9] [10]. For SN1 reactions, the nucleophile's strength is less critical because it attacks a highly reactive carbocation; thus, even weak nucleophiles (e.g., H₂O, ROH) can participate effectively [9].
Experimental Protocols and Methodologies
This section outlines general experimental workflows and key reagent solutions for investigating nucleophilic substitution pathways in a research setting.
General Workflow for Mechanistic Investigation
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents for Nucleophilic Substitution Studies
| Reagent / Material | Typical Application | Function & Rationale |
|---|---|---|
| Silver Nitrate (AgNO₃) in Ethanol | SN1 Diagnostic Test [11] | Ag⁺ precipitates halide leaving group (AgX), driving ionization of substrates capable of forming stable carbocations (e.g., tertiary, benzylic). A positive test (rapid precipitate) indicates an SN1-favorable substrate. |
| Sodium Iodide (NaI) in Acetone | SN2 Diagnostic Test (Finkelstein Test) [11] | I⁻ is a good nucleophile in the polar aprotic solvent acetone. Used to test for SN2 reactivity of primary and secondary alkyl halides, where I⁻ displaces Cl⁻ or Br⁻, forming NaX precipitate. |
| Deuterated Solvents (e.g., CD₃OD, D₂O) | Solvolysis Mechanism Tracing | Acts as both the solvent and a nucleophile. Incorporation of deuterium into products analyzed by NMR or MS provides evidence for SN1 mechanisms and carbocation rearrangement events. |
| Polar Aprotic Solvents (DMSO, DMF) | SN2 Reaction Medium [9] [10] | Solvates cations but not anions, enhancing nucleophile reactivity and favoring the bimolecular SN2 pathway. |
| Polar Protic Solvents (MeOH, EtOH, H₂O) | SN1 Reaction Medium (Solvolysis) [9] [10] | Stabilizes the carbocation intermediate and the departing leaving group via solvation, favoring the unimolecular SN1 pathway. |
| Tert-Butyl Chloride/Bromide | Model SN1 Substrate [8] [12] | A tertiary halide that readily forms a stable tertiary carbocation, serving as a standard substrate for SN1 kinetic and mechanistic studies. |
| Methyl or Primary Alkyl Halides | Model SN2 Substrate [7] | Substrates with minimal steric hindrance, serving as standard compounds for studying SN2 kinetics and stereochemistry. |
Protocol for Kinetic Order Determination
Objective: To distinguish between SN1 and SN2 mechanisms by establishing the experimental rate law.
Methodology:
- Reaction Setup: Prepare a series of reaction vessels containing the alkyl halide substrate in an appropriate solvent, maintaining a constant temperature using a thermostated bath.
- Nucleophile Variation: In one set of experiments, keep the substrate concentration constant while systematically varying the concentration of the nucleophile.
- Substrate Variation: In a parallel set of experiments, keep the nucleophile concentration constant while systematically varying the concentration of the substrate.
- Initial Rate Measurement: For each condition, monitor the reaction progress (e.g., via GC, HPLC, or conductivity measurement) to determine the initial rate of product formation or leaving group disappearance.
- Data Analysis: Plot the observed initial rate against reactant concentrations.
The strategic application of SN1 and SN2 mechanisms is fundamental to advancing organic synthesis in pharmaceutical and chemical research. A deep understanding of the interplay between substrate architecture, solvent polarity, and nucleophile potency—as detailed in this guide—enables researchers to transcend simple product prediction. This knowledge facilitates the rational design of synthetic routes, control over stereochemical outcomes, and the optimization of reaction conditions for efficiency and scalability, thereby accelerating the development of new molecular entities and therapeutic agents.
Electrophilic Addition and Elimination Reactions (E1, E2)
Understanding reaction mechanisms is a cornerstone of organic chemistry research, providing the predictive framework essential for designing synthetic routes, particularly in pharmaceutical development. Among the most critical classes of transformations are elimination and electrophilic addition reactions, which govern the interconversion between alkenes and alkyl halides or other functional groups. This guide provides an in-depth examination of the E1 and E2 elimination mechanisms and introduces the fundamentals of electrophilic addition to alkenes. Mastery of these mechanisms, including their kinetic parameters, stereoelectronic requirements, and propensity for rearrangement, enables scientists to strategically select reaction conditions to achieve desired products with high fidelity—a non-negotiable requirement in drug synthesis and development. The interplay between these pathways, often competing with nucleophilic substitution, represents a fundamental conceptual framework that researchers must navigate daily.
The E2 Elimination Mechanism
The E2 (Elimination, Bimolecular) mechanism is a concerted, single-step process in which a base abstracts a proton from the β-carbon simultaneous with the departure of a leaving group, resulting in the formation of a carbon-carbon double bond [13] [14].
Mechanistic Insights and Kinetics
The E2 mechanism is characterized by its bimolecular kinetics. The rate of the reaction is directly proportional to the concentrations of both the substrate and the base, leading to a second-order rate law: Rate = k[substrate][base] [13] [15]. This was established through classical kinetic experiments that measure how reaction rate varies with reactant concentrations.
A defining feature of the E2 mechanism is its stereochemical requirement. The reaction proceeds preferentially when the proton being abstracted and the leaving group are anti-periplanar, meaning they lie in the same plane but on opposite sides (approximately 180° dihedral angle) [13] [14]. This geometry allows the filled C-H σ-orbital to optimally overlap with the empty σ* antibonding orbital of the C-Leaving Group bond, facilitating efficient concurrent bond breaking and π-bond formation.
Diagram 1: The Concerted E2 Mechanism Pathway.
Experimental Protocol for Kinetic Study of an E2 Reaction
Objective: To determine the order of reaction with respect to the base concentration for the elimination of 1-bromopropane with sodium methoxide.
Materials:
- 1-bromopropane (substrate)
- Anhydrous methanol (solvent)
- Sodium methoxide base solution (in methanol, various concentrations: 0.05 M, 0.10 M, 0.15 M)
- Gas chromatograph (GC) or HPLC equipped with a UV-vis detector
- Heated water bath or reactor block maintained at 50°C
- Pipettes and volumetric flasks
Procedure:
- Prepare a stock solution of 1-bromopropane (0.10 M) in anhydrous methanol.
- Into each of three reaction vials, add 10 mL of the sodium methoxide solutions of different concentrations (0.05 M, 0.10 M, 0.15 M). Cap the vials to prevent moisture absorption.
- Pre-equilibrate all vials in the 50°C water bath for 5 minutes.
- Rapidly add 1.0 mL of the 1-bromopropane stock solution to each vial, starting a timer, and mix thoroughly. The final concentration of 1-bromopropane is constant in all reactions.
- At regular time intervals (e.g., 5, 10, 15, 20 minutes), withdraw a 1.0 mL aliquot from each vial and quench immediately in 1.0 mL of a 1M HCl solution to neutralize the base and stop the reaction.
- Analyze each quenched sample via GC/HPLC to determine the concentration of the remaining 1-bromopropane or the formed propene.
Data Analysis: Plot the concentration of 1-bromopropane versus time for each base concentration. The initial rates of reaction can be determined from the slope of the tangent at t=0 for each plot. A plot of the logarithm of the initial rate versus the logarithm of the base concentration will yield a straight line whose slope is the order of the reaction with respect to the base. A slope of ~1 confirms the first-order dependence, supporting the bimolecular E2 mechanism [13].
The E1 Elimination Mechanism
The E1 (Elimination, Unimolecular) mechanism is a two-step, ionic process that proceeds via a carbocation intermediate. It competes directly with the SN1 substitution reaction [16].
Mechanistic Insights and Kinetics
The E1 mechanism is characterized by unimolecular kinetics, where the rate of the reaction depends only on the concentration of the substrate and is independent of the base concentration: Rate = k[substrate] [16] [15]. This is the hallmark of a mechanism where the first step is rate-determining.
The mechanism unfolds in two distinct steps:
- Ionization (Rate-Determining Step): The leaving group departs spontaneously, forming a carbocation intermediate. This slow step governs the overall reaction rate.
- Deprotonation: A base removes a proton from the β-carbon adjacent to the carbocation, and the electrons form the new C-C π-bond.
The stability of the carbocation intermediate is the primary factor influencing the reaction rate. The stability order is methyl < primary < secondary < tertiary [16] [17] [18]. This is due to hyperconjugation, where electrons from adjacent C-H or C-C σ-bonds donate electron density into the empty p-orbital of the carbocation, and the inductive effect, where alkyl groups, being weakly electron-donating, help to stabilize the positive charge [17] [18].
Diagram 2: The Stepwise E1 Mechanism Pathway.
Experimental Protocol for Demonstrating E1 Kinetics and Carbocation Rearrangement
Objective: To observe unimolecular kinetics and a carbocation rearrangement product in the acid-catalyzed dehydration of 3,3-dimethyl-2-butanol.
Materials:
- 3,3-dimethyl-2-butanol (substrate)
- 85% Phosphoric acid (H₃PO₄) or concentrated sulfuric acid (H₂SO₄)
- Distillation apparatus
- Gas chromatograph-Mass spectrometer (GC-MS)
- NMR spectrometer ([^1]H, [^13]C)
- Separatory funnel
Procedure:
- In a round-bottom flask, add 10.0 g of 3,3-dimethyl-2-butanol and 5 mL of 85% phosphoric acid.
- Assemble the flask for simple distillation and gently heat the mixture. The alkene products will distill over as they are formed.
- Collect the distillate in an ice-cooled receiver. Continue distillation until no more organic product is collected.
- Transfer the distillate to a separatory funnel, separate the organic layer, and dry it over anhydrous magnesium sulfate.
- Analyze the crude product mixture by GC-MS to determine the number and identity of isomeric alkenes.
- Purify the product via distillation or preparative GC and analyze the major product by NMR spectroscopy.
Expected Observations and Data Analysis: The starting alcohol is secondary. A classic 1,2-alkyl shift of a methyl group will convert the initial secondary carbocation into a more stable tertiary carbocation. The major product identified by GC-MS and NMR will be 2,3-dimethyl-2-butene, not the unrearranged 3,3-dimethyl-1-butene. This result provides direct evidence for the formation of a planar carbocation intermediate that can rearrange before losing a proton. The unimolecular nature of the rate-determining step can be confirmed in a separate experiment by showing that the reaction rate is unchanged when the concentration of the acid catalyst (the proton source for the leaving group) is varied [16] [18].
Direct Comparison of E1 and E2 Reactions
A side-by-side comparison of the E1 and E2 mechanisms reveals critical distinctions that guide reaction selection.
Table 1: Comparative Analysis of E1 and E2 Elimination Mechanisms
| Feature | E1 Mechanism | E2 Mechanism |
|---|---|---|
| Molecularity | Unimolecular [16] [15] | Bimolecular [13] [15] |
| Rate Law | Rate = k[substrate] [16] [15] | Rate = k[substrate][base] [13] [15] |
| Mechanism | Two-step (carbocation intermediate) [16] | One-step (concerted) [13] |
| Base Strength | Weak base (often the solvent) [16] [15] | Strong base required [13] [15] |
| Substrate Reactivity | 3° > 2° >> 1° (driven by carbocation stability) [16] [17] | 3° > 2° > 1° (influenced by sterics and L.G. ability) [15] |
| Stereochemistry | Not stereospecific; planar carbocation intermediate [16] | Anti-periplanar requirement; stereospecific [13] [14] |
| Competing Reaction | SN1 [16] | SN2 [15] |
| Rearrangements | Common (via carbocation) [16] [18] | Never [15] |
The following decision pathway synthesizes this information into a strategic flowchart for researchers:
Diagram 3: Strategic Selection Between E1 and E2 Pathways.
The products of elimination reactions, alkenes, are pivotal synthetic intermediates. Their most characteristic reactions are electrophilic additions, where the electron-rich π-bond acts as a nucleophile [19].
The general mechanism involves two steps:
- The π-electrons attack an electrophile (E⁺), forming a new C-E bond and a carbocation intermediate.
- A nucleophile (Nu⁻) attacks the carbocation, forming the second new C-Nu bond.
A foundational rule governing the regiochemistry of these reactions is Markovnikov's Rule: in the addition of a protic acid HX to an unsymmetrical alkene, the hydrogen adds to the less substituted carbon, and the halide adds to the more substituted carbon [17] [18]. The underlying reason is stability; this pathway occurs because it proceeds through the more stable carbocation (e.g., tertiary vs. secondary, secondary vs. primary) [17]. A critical caveat for researchers is that these carbocation intermediates are susceptible to rearrangements via hydride or alkyl shifts, potentially leading to unexpected products and complicating reaction outcomes in complex synthesis [18].
Table 2: Research Reagent Solutions for Mechanism Elucidation
| Reagent / Material | Function in Experimental Protocol |
|---|---|
| Sodium Methoxide (NaOCH₃) | Strong base used to promote E2 elimination in aprotic solvents [13]. |
| Phosphoric Acid (H₃PO₄) | A strong, non-nucleophilic acid used to protonate alcohols, creating a good leaving group (H₂O) for E1 dehydration [16]. |
| Deuterated Solvents (e.g., CDCl₃) | NMR-active solvents for product characterization; deuterium labeling can be used to probe mechanism (e.g., isotope effects) [13] [14]. |
| Gas Chromatograph-Mass Spectrometer (GC-MS) | Essential analytical instrument for separating and identifying mixture components (e.g., isomeric alkenes, rearranged products) [18]. |
| Polar Aprotic Solvent (e.g., DMSO) | Enhances base reactivity by solvating cations but not anions, favoring E2 over SN2 with strong nucleophiles [13]. |
| Polar Protic Solvent (e.g., H₂O, EtOH) | Ionizing solvent that stabilizes carbocation intermediates and leaving groups, favoring E1 and SN1 mechanisms [16] [17]. |
Reactive intermediates are short-lived, high-energy molecules formed during the stepwise conversion of reactants into products in organic reactions [20]. Their transient nature, often lasting mere seconds or less, makes them challenging to isolate and study directly. However, understanding their formation, structure, and stability is fundamental to elucidating reaction mechanisms and enables chemists to design more efficient synthetic pathways, optimize yields, and develop new catalytic processes [20] [21]. This guide provides an in-depth examination of three pivotal carbon-centered intermediates—carbocations, carbanions, and free radicals—framed within the context of modern organic chemistry research and its applications in fields such as drug development.
Structural Properties and Stability Trends
The reactivity and lifetime of carbocations, carbanions, and free radicals are governed by their electronic structures and the stability imparted by their molecular environment.
Carbocations
A carbocation is a positively charged species featuring a trivalent, electron-deficient carbon atom with an empty p-orbital [17]. This structure confers certain characteristics:
- Geometry: sp2 hybridized and trigonal planar, allowing for nucleophilic attack from either face of the empty p-orbital [17].
- Electron Configuration: Possesses only six valence electrons, making it a potent Lewis acid [17].
Stability Factors for Carbocations [17]:
- Inductive/Hyperconjugative Stabilization: Electron donation from adjacent alkyl groups through sigma bonds increases stability. The stability order is: methyl < primary < secondary < tertiary.
- Resonance Stabilization: Delocalization of the positive charge through adjacent pi systems (e.g., allylic and benzylic carbocations) greatly enhances stability. The trityl carbocation (Ph3C+) is so stable it forms crystalline salts [17].
- Adjacent Lone Pairs: A heteroatom (e.g., oxygen) with a lone pair adjacent to the carbocation center can provide stability through resonance, so long as the atom can accommodate the positive charge without violating the octet rule.
Carbanions
A carbanion is a negatively charged species where the carbon atom possesses a lone pair of electrons and a formal charge of -1 [21]. Key properties include:
- Geometry: Can be sp3 hybridized (tetrahedral) or sp2 hybridized (trigonal planar), influenced by the attached groups and the possibility of resonance [21].
- Electron Configuration: The carbon atom has eight valence electrons, making it electron-rich and nucleophilic.
Stability Factors for Carbanions [22] [23]:
- Inductive Effect: The negative charge is stabilized by adjacent electron-withdrawing groups (e.g., -NO2, -CN, carbonyl groups).
- Hybridization: Stability increases with greater s-character of the orbital bearing the lone pair (sp > sp2 > sp3).
- Alkyl Substitution: Stability decreases with increasing alkyl substitution due to the electron-donating inductive effect of alkyl groups, which intensifies the charge density. Thus, the stability order is: primary > secondary > tertiary [22] [23].
- Resonance Stabilization: As with carbocations, delocalization of the negative charge into adjacent pi systems (e.g., enolates) dramatically increases stability.
Free Radicals
A free radical is a neutral, electron-deficient species featuring an unpaired electron on carbon [23] [21].
- Geometry: Can be sp2 or sp3 hybridized, but is often nearly planar to facilitate delocalization [21].
- Electron Configuration: The carbon has seven valence electrons, making it highly reactive.
Stability Factors for Free Radicals [22] [23]:
- Inductive/Hyperconjugative Stabilization: Similar to carbocations, stability increases with greater alkyl substitution: methyl < primary < secondary < tertiary.
- Resonance Stabilization: Delocalization of the unpaired electron (e.g., allylic or benzylic radicals) significantly enhances stability.
Table 1: Comparative Stability Trends of Reactive Intermediates Based on Alkyl Substitution
| Intermediate Type | Charge | Stability Order (by substitution) | Key Stabilizing Factor |
|---|---|---|---|
| Carbocation | Positive | Tertiary > Secondary > Primary > Methyl | Hyperconjugation & Resonance |
| Carbanion | Negative | Primary > Secondary > Tertiary | Inductive effect (EWGs) |
| Free Radical | Neutral | Tertiary > Secondary > Primary > Methyl | Hyperconjugation & Resonance |
Table 2: Impact of Resonance on Intermediate Stability
| Intermediate Type | Non-Resonance Stabilized Example | Resonance Stabilized Example | Stability Enhancement |
|---|---|---|---|
| Carbocation | CH3CH2+ (Primary) | Allyl Cation (CH2=CH-CH2+) | Very Large |
| Carbanion | CH3CH2- (Primary) | Enolate (CH2=CHO-) | Very Large |
| Free Radical | CH3CH2* (Primary) | Allyl Radical (CH2=CH-CH2*) | Very Large |
Experimental and Computational Methodologies
The fleeting nature of reactive intermediates demands sophisticated techniques for their detection and characterization. Advances in spectroscopy, electrochemistry, and computational modeling have provided powerful tools for directly observing these species and validating their roles in reaction mechanisms.
Spectroscopic Techniques
- Nuclear Magnetic Resonance (NMR) Spectroscopy: Stopped-flow and high-field NMR can detect intermediates with moderately short lifetimes. Specific chemical shifts for protons and carbon atoms in the intermediate provide structural fingerprints [21].
- Infrared (IR) Spectroscopy: Time-resolved IR spectroscopy, often triggered by a laser pulse, can monitor the formation and decay of intermediates by tracking characteristic vibrational frequencies of functional groups. Attenuated Total Reflectance (ATR-IR) is particularly useful for in-situ analysis [21].
- Mass Spectrometry (MS): Soft ionization techniques like Electrospray Ionization Mass Spectrometry (ESI-MS) allow for the detection of intact, charged intermediates. Coupled with fast-flow or stopped-flow reactors, MS can track reaction kinetics and identify transient species [21].
Advanced Electrochemical Analysis
A groundbreaking 2025 study demonstrated the spatio-temporal visualization of carbocation intermediates within an electrolytic cell [24]. The methodology is detailed below and summarized in the workflow diagram.
Experimental Workflow for Spatio-Temporal Visualization [24]:
- Electrochemical Generation: A custom-designed electrochemical cell generates carbocations near the anode during oxidative transformations.
- Spatio-Temporal Sampling: At precise time intervals and spatial locations (e.g., from the anode to the cathode), a micro-syringe withdraws a 10 μL aliquot from the reaction solution.
- Rapid Analysis via DESI-MS: The aliquot is immediately dispensed onto a glass slide and subjected to a spray of charged water microdroplets in a Desorption Electrospray Ionization (DESI) source. The superacidic microdroplet environment stabilizes the carbocations.
- Detection and Mapping: The stabilized ions are pneumatically propelled into the mass spectrometer for detection. By repeating this process, a contour plot is generated, visualizing the abundance and diffusion of the carbocation throughout the cell over time.
Diagram 1: Spatio-temporal visualization workflow.
This technique confirmed that carbocation abundance is highest near the anode and decreases towards the cathode, providing unprecedented insight into the dynamics of electrochemical reactions [24].
Computational Chemistry
When experimental detection is prohibitively challenging, computational methods offer a powerful alternative for predicting and characterizing intermediates.
- Density Functional Theory (DFT) Calculations: DFT is used to optimize the geometry of proposed intermediates and calculate their energies, providing a detailed reaction energy profile. Analysis of activation barriers helps identify rate-determining steps and the most favorable reaction pathway [21].
- Molecular Modeling and Dynamics: These methods provide detailed structural information, including bond lengths and angles. Calculated vibrational frequencies can be matched with experimental IR data to support the identification of an intermediate. Molecular dynamics simulations can model the behavior of intermediates in complex environments, such as enzyme active sites [21].
Table 3: Key Experimental Methods for Intermediate Analysis
| Method | Key Principle | Typical Time Resolution | Primary Information Obtained |
|---|---|---|---|
| Stopped-Flow NMR | Nuclear spin relaxation in a magnetic field | Milliseconds to seconds | Molecular structure, bonding, dynamics |
| Time-Resolved IR | Vibration of chemical bonds | Picoseconds to microseconds | Functional group identity and environment |
| DESI-MS | Mass-to-charge ratio of ions | Real-time (seconds) | Molecular mass, elemental composition |
| Electrochemical Kinetics | Reaction rate measurement | Varies with technique | Reaction order, rate constants, mechanism |
The Scientist's Toolkit: Essential Reagents and Materials
This table details key reagents and materials used in the advanced experimental detection of reactive intermediates, as exemplified by the 2025 DESI-MS study [24].
Table 4: Key Research Reagent Solutions for Intermediate Analysis
| Reagent / Material | Function in the Experiment |
|---|---|
| Reticulated Vitreous Carbon (RVC) Electrode | Serves as a high-surface-area anode for the electrochemical generation of radical cations and carbocations. |
| Custom Electrolytic Cell | A reaction vessel designed with multiple sampling ports to allow for the spatial mapping of intermediates. |
| Hamilton Syringe Needle | Enables the precise withdrawal of micro-liter volume aliquots (10 μL) from specific locations within the electrolytic cell. |
| Charged Water Microdroplets (DESI Spray) | Creates a superacidic environment at the air-water interface that stabilizes fleeting carbocations, preventing their decomposition prior to mass spectrometric detection. |
| Desorption Electrospray Ionization (DESI) Source | The interface that rapidly introduces and ionizes the sampled aliquot for transfer into the mass spectrometer. |
| High-Resolution Mass Spectrometer | Accurately measures the mass-to-charge (m/z) ratio of the ionized intermediates, allowing for their definitive identification. |
Implications for Reaction Mechanisms and Synthesis
A profound understanding of reactive intermediates directly translates to the rational design and optimization of synthetic methodologies, which is critical in pharmaceutical process chemistry.
Elucidating and Controlling Reaction Pathways
Identifying the involvement of a carbocation, for instance, immediately informs the mechanism. In nucleophilic substitution, a carbocation intermediate points to an SN1 mechanism, which has different stereochemical and kinetic implications compared to the concerted SN2 pathway [21]. This knowledge allows researchers to manipulate reaction conditions—such as solvent polarity, temperature, and catalyst design—to steer the reaction along the desired path, minimizing side reactions and improving yield [21].
Guiding Sustainable Process Development
Modern organic synthesis emphasizes sustainability. The strategic use of reactive intermediates enables more efficient, atom-economical routes to complex molecules. For example, the field of electroorganic synthesis, as highlighted in the 2025 study, uses electricity to generate intermediates like carbocations, providing a sustainable alternative to stoichiometric oxidants [24]. Furthermore, catalytic cycles in both organometallic and biocatalysis often rely on the generation and controlled reactivity of these species to achieve high selectivity and reduce waste [25] [26].
Carbocations, carbanions, and free radicals represent fundamental reactive intermediates whose properties and behaviors underpin a vast array of organic transformations. Mastery of their stability trends, coupled with modern techniques for their direct detection and computational modeling, provides researchers with a powerful framework for mechanistic analysis. As experimental methods, such as the spatio-temporal profiling demonstrated in recent research, continue to evolve, our ability to observe and manipulate these fleeting species will deepen. This ongoing advancement is essential for driving innovation in the synthesis of complex molecules, including active pharmaceutical ingredients, and for developing new, sustainable chemical processes.
Rearrangement reactions represent a fundamental class of transformations in organic chemistry where the carbon skeleton of a molecule undergoes reorganization, leading to structural isomers with distinct connectivity. Within the broader thesis of reaction mechanism research, these rearrangements provide critical insights into carbocation behavior, reaction dynamics, and stereoelectronic effects that govern molecular stability and reactivity. The Wagner-Meerwein rearrangement, first discovered in the late 19th century through the work of Georg Wagner and Hans Meerwein, stands as a paradigmatic example of such transformations [27]. Their investigations into the conversion of isoborneol to camphene revealed unexpected molecular reorganizations that challenged contemporary understanding of reaction mechanisms and ultimately led to the recognition of carbocations as genuine reaction intermediates [27] [28].
The historical significance of these rearrangements extends beyond mere academic curiosity, as they established foundational principles for understanding how cationic intermediates dictate reaction pathways in complex molecular systems. The intellectual journey from phenomenological observation to mechanistic understanding exemplifies how studying rearrangement reactions has profoundly shaped modern physical organic chemistry [27]. Within pharmaceutical research and natural product synthesis, these rearrangements frequently emerge as both challenges and opportunities—complicating predicted reaction outcomes while enabling sophisticated skeletal transformations that would otherwise be inaccessible through conventional synthetic approaches.
Fundamental Principles and Mechanisms
Carbocation Stability and Rearrangement Driving Forces
Carbocation stability serves as the primary thermodynamic driving force for molecular rearrangements, with the stability order following the established pattern: methyl < primary < secondary < tertiary < resonance-stabilized cations [29] [28]. This stability gradient creates the fundamental imperative for rearrangement—whenever a less stable carbocation can transform into a more stable carbocation through migration of an adjacent substituent, the rearrangement becomes energetically favorable [29]. The electron-deficient nature of carbocations (possessing only six electrons in their valence shell) makes them particularly susceptible to stabilization through both inductive effects from adjacent alkyl groups and hyperconjugation, wherein neighboring C-H and C-C bonds donate electron density to empty the p-orbital [29].
Beyond thermodynamic stability considerations, structural strain relief provides an additional powerful driving force for rearrangements [28]. Angle strain in small rings (particularly cyclopropane and cyclobutane systems), torsional strain in conformationally restricted systems, and steric crowding in highly substituted molecules can all provide sufficient energetic incentive to trigger skeletal reorganizations even when the carbocation stability remains unchanged [28]. In such cases, the rearrangement represents a trade-off between increased carbocation stability and decreased ring strain, with the net energetic benefit determining whether the rearrangement occurs. This dual consideration of electronic and strain effects provides a more complete framework for predicting rearrangement outcomes in complex bicyclic and polycyclic systems commonly encountered in terpenoid natural products [28].
The Molecular Mechanism of Hydride Shifts
Hydride shifts constitute one of the most prevalent forms of carbocation rearrangements, characterized by the migration of a hydrogen atom with its bonding electron pair from one carbon to an adjacent electron-deficient carbon [29]. The mechanism proceeds through a concerted process in which the C-H bond aligns with the empty p-orbital of the carbocation, enabling the bonding electrons to bridge the gap between the two carbon centers in a transition state that resembles a relay race baton pass [29]. This [1,2]-sigmatropic shift occurs with stereochemical retention at the migrating hydrogen and proceeds suprafacially, meaning all bond formations and breaking occur on the same face of the molecular system [27] [28].
The kinetics of hydride shifts are remarkably facile, with many occurring spontaneously at temperatures as low as -120°C, indicating exceptionally low energy barriers for these processes [27]. The rearrangement rate depends critically on the structural alignment between the donating C-H bond and the accepting carbocation center, with optimal geometry occurring when the three centers (migration origin, migrating hydrogen, and migration terminus) can achieve a colinear arrangement that maximizes orbital overlap throughout the transition state [29]. This geometric requirement explains why certain rigid molecular frameworks exhibit particularly efficient hydride shifts while more flexible systems may experience competitive side reactions.
Table 1: Comparative Analysis of Hydride Shift Energetics in Different Structural Contexts
| Structural Context | Energy Barrier | Rate at 25°C | Driving Force |
|---|---|---|---|
| Secondary to tertiary | Low | Very fast | ~15 kcal/mol stability increase |
| Primary to secondary | Moderate | Fast | ~10-12 kcal/mol stability increase |
| Secondary to resonance-stabilized | Very low | Instantaneous | >20 kcal/mol stability increase |
| Strain-relieving shifts | Variable | Dependent on strain relief | 5-15 kcal/mol strain energy relief |
Wagner-Meerwein Rearrangements: Scope and Variations
Wagner-Meerwein rearrangements encompass a broader class of 1,2-shifts that includes not only hydride migrations but also alkyl and aryl group transfers [27]. These reactions follow the same fundamental pattern—a group migrates from one carbon to an adjacent carbocation center with its bonding electrons—but differ in the nature of the migrating species. The pericyclic description classifies these as cationic [1,2]-sigmatropic rearrangements with the Woodward-Hoffmann symbol [ω0s + σ2s], indicating a thermally allowed process that conserves orbital symmetry throughout the transformation [27].
The migrating aptitude of different groups follows the general trend: H > aryl > alkyl, though this ordering can be influenced by stereoelectronic factors, steric constraints, and the specific geometry of the molecular framework [27] [28]. In bicyclic terpene systems, these rearrangements often proceed through a series of consecutive shifts that dramatically alter the carbon skeleton, as exemplified by the conversion of isoborneol to camphene that originally captured Wagner's attention [27]. The related Nametkin rearrangement, which involves specific methyl group migrations in terpenes, represents a specialized subclass of Wagner-Meerwein transformations that highlight the sensitivity of these processes to subtle structural features [27].
Experimental Methodologies and Protocols
Standard Laboratory Protocol for Observing Hydride Shifts
The following experimental procedure provides a reliable method for demonstrating carbocation rearrangements in a pedagogical or research setting, adapted from established educational experiments [27] [29]:
Reaction Setup and Execution:
- Substrate Preparation: Dissolve 2.0 mmol of a secondary alcohol substrate (e.g., 3-methyl-2-butanol) in 10 mL of glacial acetic acid in a 50 mL round-bottom flask equipped with a magnetic stir bar.
- Acid Initiation: Slowly add 5 mL of concentrated sulfuric acid dropwise with continuous stirring while maintaining the reaction temperature between 0-5°C using an ice bath.
- Reaction Monitoring: Allow the reaction mixture to warm to room temperature and stir for 2 hours, monitoring by TLC (9:1 hexane:ethyl acetate) to confirm consumption of the starting material.
- Workup Procedure: Carefully pour the reaction mixture onto 20 g of crushed ice, then extract with three 15 mL portions of dichloromethane.
- Purification: Combine the organic extracts and wash successively with saturated sodium bicarbonate solution (until neutral), brine, then dry over anhydrous magnesium sulfate.
- Product Analysis: Filter and concentrate under reduced pressure to obtain the crude product, which can be analyzed by NMR spectroscopy to detect rearrangement through comparison with authentic standards.
Key Analytical Signatures:
- ¹H NMR: Disappearance of signals characteristic of the starting material carbocation and emergence of new signals corresponding to the rearranged skeleton.
- Product Distribution: Comparison of products from isomeric starting materials (e.g., 2-methyl-2-butanol vs. 3-methyl-2-butanol) showing identical product distributions confirms the rearrangement process [27] [29].
Advanced Protocol: Biomimetic Synthesis of Clovane-Type Terpenoids
Recent methodological advances have demonstrated the utility of Wagner-Meerwein rearrangements in complex natural product synthesis. The following protocol for the biomimetic synthesis of clovane-type terpenoids illustrates the application of these rearrangements in sophisticated target-oriented synthesis [30]:
Epoxide Initiation and Cascade Rearrangement:
- Substrate Activation: Dissolve 1.0 mmol of (-)-caryophyllene β-oxide in 20 mL of anhydrous dichloromethane under nitrogen atmosphere.
- Lewis Acid Catalysis: Add 0.1 mmol of tin(IV) chloride dropwise at -78°C with vigorous stirring.
- Cascade Initiation: Warm the reaction mixture gradually to 0°C over 30 minutes, allowing the epoxide opening to trigger a series of transannular cyclizations and Wagner-Meerwein rearrangements.
- Reaction Quenching: Add 1 mL of triethylamine to quench the Lewis acid, then dilute with 30 mL of ethyl acetate.
- Intermediate Trapping: Wash the organic layer with water and brine, then dry over sodium sulfate.
- Product Diversification: Concentrate under reduced pressure and subject the crude product to nucleophilic trapping conditions (methanol, water, or ammonia) to yield functionalized clovane derivatives with characteristic C2 and C9 oxygenation patterns [30].
Strategic Considerations:
- This biomimetic approach constructs the complex tricyclo[6.3.1.0¹,⁵]dodecane core characteristic of clovane terpenoids in a single synthetic operation.
- The cascade sequence proceeds through a bridgehead carbocation intermediate that undergoes precisely orchestrated Wagner-Meerwein rearrangement to establish the clovane skeleton [30].
- Modifying the nucleophilic trapping agents enables diversification of the oxygenation pattern, providing access to multiple natural product analogues from a common intermediate.
Visualization of Reaction Pathways and Mechanisms
Diagram 1: Hydride shift mechanism in SN1 reactions
Diagram 2: Biosynthetic pathway to clovane terpenoids
Research Reagent Solutions and Essential Materials
Table 2: Essential Research Reagents for Investigating Carbocation Rearrangements
| Reagent/Catalyst | Function in Rearrangement Studies | Application Examples |
|---|---|---|
| Concentrated H₂SO₄ | Brønsted acid catalyst for carbocation generation | Protonation of alcohols to form carbocation intermediates in educational demonstrations [29] |
| SnCl₄ | Lewis acid catalyst for epoxide opening | Initiation of cascade rearrangements in biomimetic terpenoid synthesis [30] |
| Polyphosphoric Acid | Mild acid catalyst for sensitive substrates | Promotion of Wagner-Meerwein rearrangements without decomposition of products [27] |
| Anhydrous Organic Solvents (CH₂Cl₂, EtOAc) | Inert reaction media for cationic processes | Maintaining carbocation integrity by excluding nucleophiles during rearrangement [30] |
| Deuterated Solvents (CDCl₃, DMSO-d₆) | NMR analysis of rearrangement mechanisms | Monitoring carbocation rearrangements in situ using low-temperature NMR techniques |
Applications in Complex Molecule Synthesis and Drug Development
The strategic implementation of Wagner-Meerwein rearrangements has enabled remarkable advances in complex molecule synthesis, particularly within the realm of terpenoid natural products possessing intricate polycyclic architectures. Contemporary research has demonstrated the power of these rearrangements in achieving collective total syntheses of structurally related natural products from common synthetic intermediates [30]. The recent enantioselective total synthesis of rumphellclovane E and sarinfacetamides A and B exemplifies this approach, where a biomimetic Wagner-Meerwein rearrangement constructs the characteristic tricyclo[6.3.1.0¹,⁵]dodecane core shared by these clovane-type terpenoids [30].
In pharmaceutical research, the ability to rapidly generate molecular complexity through cascade rearrangement sequences offers significant strategic advantages. The synthesis of sarinfacetamides is particularly noteworthy, as these compounds exhibit unique biological activity promoting ConA-induced T lymphocyte proliferation, suggesting potential immunomodulatory applications [30]. The biosynthetic paradigm, wherein caryophyllene-type terpenoids undergo enzyme-initiated epoxide opening followed by Wagner-Meerwein rearrangement to establish the clovane skeleton, provides a blueprint for biomimetic synthetic approaches that maximize efficiency while minimizing functional group manipulations [30]. This strategy has enabled the synthesis of structural analogues containing diverse functionalization patterns, including furan moieties, expanding the accessible chemical space for structure-activity relationship studies in drug discovery programs.
The development of novel synthetic methodologies continues to expand the applications of Wagner-Meerwein rearrangements in complex synthesis contexts. Recent innovations include domino Michael-aldol sequences that establish key bicyclo[3.3.1]nonane intermediates, which subsequently undergo Wagner-Meerwein rearrangements to generate architecturally complex terpenoid frameworks [30]. These methodologies address longstanding challenges in terpenoid synthesis, particularly the introduction of diverse side chain moieties at the C4 position—a structural feature present in several biologically active clovane derivatives. The modularity of this approach enables systematic variation of side chain structures, facilitating the exploration of structure-activity relationships while maintaining the complex core architecture essential for biological activity.
Quantitative Data and Comparative Analysis
Table 3: Kinetic and Thermodynamic Parameters for Representative Rearrangements
| Rearrangement Type | Activation Energy (kcal/mol) | Temperature Range | Rate Enhancement vs. Primary Carbocation |
|---|---|---|---|
| Primary to Secondary | 12-15 | -20°C to 25°C | 10²-10³ |
| Secondary to Tertiary | 8-12 | -120°C to 0°C | 10⁴-10⁶ |
| Alkyl vs Hydride Migration | ΔΔG‡ = 3-5 | Comparable conditions | Hydride 10-100x faster |
| Strain-Driven Shifts | 5-10 | -78°C to 25°C | Variable based on ring strain |
The quantitative analysis of rearrangement kinetics reveals consistent patterns across diverse structural families. Wagner-Meerwein rearrangements typically proceed with activation barriers of 8-15 kcal/mol, making them exceptionally fast processes that often compete effectively with direct nucleophilic capture of the initially formed carbocation [27] [28]. The temperature dependence of these rearrangements is notably shallow, with many proceeding efficiently at cryogenic temperatures (-120°C), indicating minimal entropic barriers and highly ordered, concerted transition states [27]. This low temperature capability enables the spectroscopic observation of rearrangement intermediates using advanced techniques such as low-temperature NMR, providing direct structural evidence for the proposed mechanisms.
Comparative analysis of migrating group aptitude reveals that hydride shifts generally proceed 10-100 times faster than comparable alkyl migrations, reflecting the greater nucleophilicity and lower steric demand of hydrogen versus carbon migration [28]. However, this intrinsic preference can be overridden by stereoelectronic factors, particularly in conformationally constrained polycyclic systems where geometric alignment between the migrating group and the empty p-orbital dictates rearrangement efficiency. In systems where multiple rearrangement pathways are geometrically accessible, the relative rates determine the product distribution, with faster hydride shifts often dominating over thermodynamically favored alkyl migrations when the two processes are competitive.
Pericyclic reactions represent a fundamental class of organic reactions characterized by a concerted mechanism, proceeding through a cyclic transition state without the formation of intermediates [31]. In these processes, the reorganization of bonding electrons occurs in a single, coordinated step, governed by the symmetry properties of the involved molecular orbitals [31]. This concerted nature, with bond formation and breakage occurring simultaneously, distinguishes pericyclic reactions from their ionic or radical counterparts and imbues them with high stereospecificity, a hallmark feature extensively documented in research [32].
The theoretical framework for understanding these reactions was profoundly advanced by the Woodward-Hoffmann rules, which established the principle of the conservation of orbital symmetry [31]. This principle dictates that for a pericyclic reaction to be thermally allowed, the molecular orbitals of the reactants must correlate symmetrically with those of the products in their ground states [31]. Several equivalent theoretical approaches, including frontier molecular orbital (FMO) theory and the aromatic transition state theory, provide complementary insights. The latter posits that the minimum energy transition state for a pericyclic process must be aromatic, with Hückel topology favored for (4n + 2)-electron systems and Möbius topology for 4n-electron systems [31]. These reactions are equilibrium processes, though they can be driven to completion when products are significantly more stable [31].
The study of pericyclic reactions is crucial in organic chemistry research, particularly in synthetic methodology development and understanding biosynthetic pathways. Their predictable stereochemical outcomes make them indispensable for constructing complex molecular architectures with precise stereocontrol, a critical requirement in drug development for crafting molecules with specific biological activities.
Major Pericyclic Reaction Classes
Cycloadditions
Cycloaddition reactions involve the combination of two or more π-systems to form a cyclic adduct with new σ-bonds, typically denoted as [i+j] where i and j represent the number of π-electrons contributed by each component [32] [33]. The most prominent cycloaddition is the Diels-Alder reaction, a [4π+2π] cyclization between a diene and a dienophile [32]. This reaction exhibits remarkable stereospecificity, maintaining the configuration of substituents from reactants to products [32]. For instance, trans-configured substituents on the dienophile remain trans in the cycloadduct [32].
The orientation of substituents in the bicyclic products leads to endo or exo diastereomers, with the endo product (where the substituent is oriented cis to the longest or more unsaturated bridge) often favored under kinetic control [32]. Intramolecular Diels-Alder reactions are particularly powerful for constructing complex polycyclic systems, forming two new rings simultaneously—one from the cycloaddition itself and another from the conformational constraints of the linking chain [32]. Other cycloaddition variants include [6+4] cyclizations and photochemical [2+2] cycloadditions, the latter proceeding with different stereochemical rules due to its photochemical nature [32].
Electrocyclic Reactions
Electrocyclic reactions involve the concerted cyclization of a conjugated π-system, converting a π-bond to a ring-forming σ-bond, or the reverse ring-opening process [32]. These transformations exhibit distinctive stereospecificity dependent on the number of π-electrons and whether the reaction occurs under thermal or photochemical conditions [32].
A classic example is the thermal ring closure of trans,cis,trans-2,4,6-octatriene to cis-5,6-dimethyl-1,3-cyclohexadiene [32]. The stereochemical outcome is rigorously determined by the conrotatory (rotation in the same direction) or disrotatory (rotation in opposite directions) motion of the terminal orbitals during ring closure, which is governed by the Woodward-Hoffmann rules [33]. The reverse process, electrocyclic ring opening, is often favored when it relieves ring strain, such as in the opening of cyclobutenes to conjugated butadienes [32].
Sigmatropic Rearrangements
Sigmatropic rearrangements involve the migration of a σ-bond flanked by π-systems to a new position with concomitant reorganization of the π-bonds [32]. These reactions are described by bracketed numbers [i,j] indicating the relative distance each end of the σ-bond has moved [32].
Common sigmatropic shifts include [1,5] hydrogen shifts, which convert unstable allene systems to conjugated trienes [32]. Notably, [1,3] hydrogen shifts are not typically observed in thermal pericyclic reactions, with preference for [1,5] shifts in appropriate systems [32]. The [3,3] sigmatropic rearrangements encompass the Cope rearrangement of 1,5-dienes and the Claisen rearrangement of allyl vinyl ethers [32]. These reactions are stereospecific and highly valuable in synthetic applications, particularly the oxy-Cope variant where a hydroxyl substituent at the central carbon accelerates the rearrangement when converted to its alkoxide salt [32]. The Claisen rearrangement of allyl phenyl ethers to ortho-allyl phenols demonstrates its synthetic utility, with the initial cyclohexadienone product rapidly tautomerizing to regain aromatic stabilization [32].
Ene Reactions
The ene reaction joins a double or triple bond (enophile) to an alkene with a transferable allylic hydrogen (ene), characterized by redistribution of three pairs of bonding electrons [32]. The reverse process is termed a retro ene reaction [32]. These reactions are favored when the enophile is electrophilic, and they can proceed intermolecularly or intramolecularly, with the latter capable of forming new rings [32]. While hydrogen is the most commonly transferred atom, other groups can participate in ene-like transformations [32]. Lewis acid catalysis can significantly enhance the reaction rate, as seen in the Prins reaction where simple aldehydes react with alkenes to form allylic alcohols, 1,3-diols, or 1,3-dioxanes [32].
Table 1: Major Pericyclic Reaction Classes and Their Characteristics
| Reaction Class | Key Transformation | Electron Count | Stereochemical Feature |
|---|---|---|---|
| Cycloaddition | Two π-systems form cyclic adduct | Varied ([4+2], [2+2], etc.) | Stereospecific; endo/exo selectivity |
| Electrocyclic | Ring closure/opening of conjugated π-system | 4n or 4n+2 π-electrons | Conrotatory/Disrotatory ring closure |
| Sigmatropic | σ-bond migration with π-bond reorganization | [i,j] numbering system | Stereospecific shift |
| Ene Reaction | Allylic hydrogen transfer to enophile | Six-electron reorganization | Can be intramolecular for ring formation |
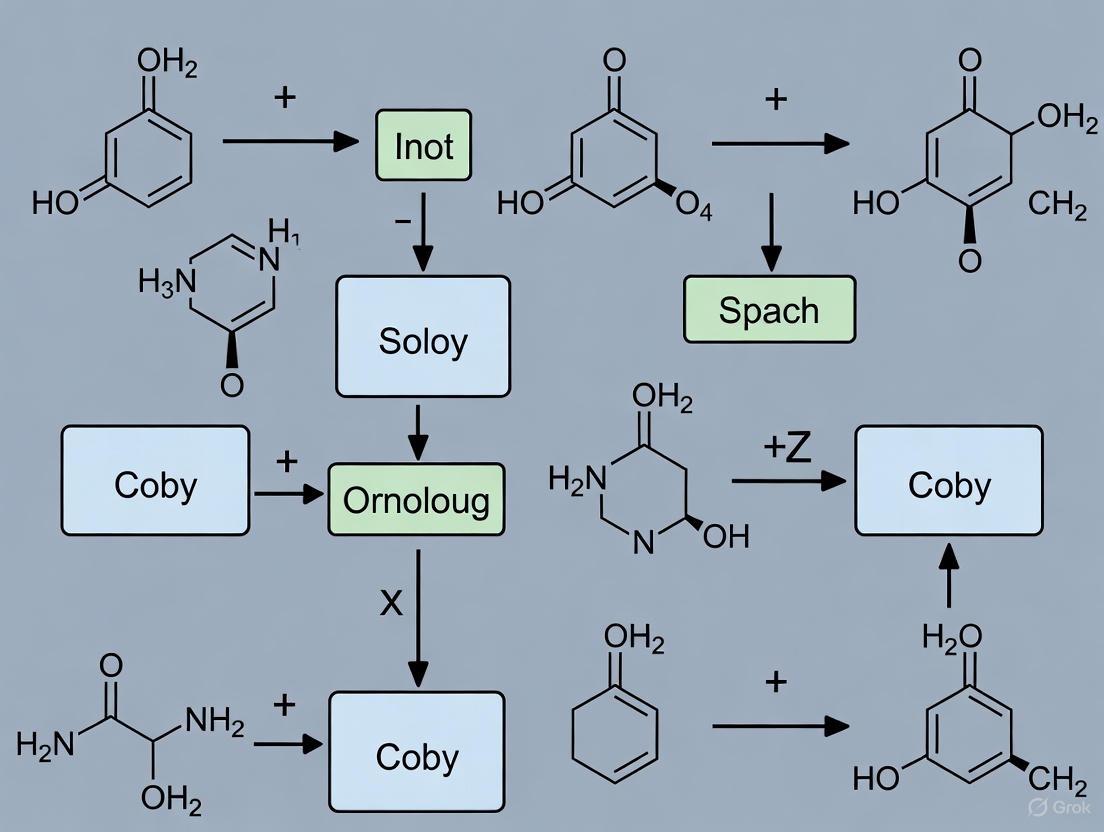
Enzymatic Catalysis of Pericyclic Reactions
Biological Significance and the PchB Enzyme Case Study
Pericyclic reactions occur in several biological processes, highlighting their fundamental importance in biochemistry [31]. Notable examples include the Claisen rearrangement of chorismate to prephenate in almost all prototrophic organisms, [1,5]-sigmatropic shifts in the transformation of precorrin-8x to hydrogenobyrinic acid, and the non-enzymatic, photochemical electrocyclic ring opening followed by a (1,7) sigmatropic hydride shift in vitamin D synthesis [31].
The isochorismate-pyruvate lyase (PchB) from Pseudomonas aeruginosa provides a compelling case study of enzymatic pericyclic catalysis [34]. This enzyme is involved in siderophore biosynthesis, specifically converting isochorismate to salicylate and pyruvate through a concerted but asynchronous [1,5]-sigmatropic shift with quantitative hydrogen transfer from C2 to C9, as confirmed by NMR and computational studies [34]. Remarkably, PchB also exhibits catalytic promiscuity, performing a non-physiological role as a chorismate mutase (a Claisen rearrangement) with considerably lower catalytic efficiency (k~cat~/K~m~ = 1.96×10^2^ M^−1^s^−1^) compared to its primary lyase activity (k~cat~/K~m~ = 4.11×10^4^ M^−1^s^−1^) [34].
Structural Insights and Catalytic Mechanisms
PchB shares approximately 20% sequence similarity and is a structural homolog of the AroQ class chorismate mutases, such as E. coli chorismate mutase (EcCM) [34]. Structural analyses reveal that PchB conserves only five of the eight charged/polar amino acids responsible for binding the transition state analog in EcCM, which may contribute to its catalytic promiscuity [34]. A significant structural difference exists between the apo-form and ligand-bound forms of PchB: the active site loop between helix one and helix two is disordered in the apo-structure but becomes fully ordered when pyruvate or both pyruvate and salicylate are bound, suggesting an induced-fit mechanism with open and closed states for substrate entry and catalysis, respectively [34].
Two primary hypotheses have emerged to explain enzymatic catalysis of pericyclic reactions:
Electrostatic Transition State Stabilization: This model emphasizes the role of positively charged active site residues in stabilizing developing negative charges during bond reorganization [34]. Mutational studies support this hypothesis; for example, changing Arg90 to citrulline in Bacillus subtilis chorismate mutase (BsCM) decreases catalytic efficiency by at least three orders of magnitude [34].
Near Attack Conformation (NAC): This theory proposes that enzymes pre-organize substrates into reactive conformations where reacting atoms are within van der Waals contact distance and properly aligned for bond formation [34]. Quantum mechanical/molecular mechanical molecular dynamics simulations (QM/MM-MD) suggest that once this NAC is formed, the reaction proceeds spontaneously without further electrostatic stabilization [34].
These mechanisms are not necessarily mutually exclusive, and current research suggests that enzymatic pericyclic catalysis likely arises from unique dynamic properties tuned to promote specific chemistries, rather than from static active site features alone [34].
Table 2: Kinetic Parameters and Structural Features of Selected Pericyclic Enzymes
| Enzyme | Reaction Catalyzed | k~cat~/K~m~ (M^−1^s^−1^) | Key Structural Features |
|---|---|---|---|
| PchB (lyase activity) | [1,5]-sigmatropic shift | 4.11 × 10^4^ | Homolog of AroQ class; conserved Lys42; disordered-to-ordered loop transition |
| PchB (mutase activity) | Claisen rearrangement | 1.96 × 10^2^ | Same active site as lyase activity but lower efficiency |
| EcCM | Claisen rearrangement | 2.4 × 10^5^ | Intertwined dimer; transition state stabilized by arginine residues from both monomers |
| BsCM | Claisen rearrangement | 1 × 10^6^ | Trimeric pseudo α/β-barrel; critical Arg90 for catalysis |
| mMjCM | Claisen rearrangement | 1.9 × 10^4^ | Engineered monomer; molten globule without ligand |
Experimental Methodologies and Research Tools
Kinetic Analysis of Pericyclic Enzymes
Detailed kinetic characterization provides fundamental insights into enzymatic pericyclic reactions. For PchB, the kinetic parameters for both its physiological ([1,5]-sigmatropic shift) and promiscuous (Claisen rearrangement) activities were determined using spectrophotometric assays monitoring substrate depletion or product formation [34]. The significant difference in catalytic efficiency (k~cat~/K~m~) between the two activities—two orders of magnitude higher for the lyase function—suggests evolutionary optimization for the physiological reaction despite structural similarities with chorismate mutases [34].
Protocol for Determining Kinetic Parameters:
- Enzyme Purification: Recombinant PchB is expressed in E. coli and purified using affinity chromatography followed by size-exclusion chromatography [34].
- Assay Conditions: Reactions are typically conducted in buffered aqueous solutions (e.g., 50 mM HEPES, pH 7.5) at 25°C [34].
- Substrate Preparation: Isochorismate is enzymatically synthesized from chorismate or obtained commercially. Substrate concentrations should span a range above and below the expected K~m~ [34].
- Reaction Monitoring: For the lyase reaction, monitor the decrease in isochorismate absorbance at 250 nm or the increase in salicylate fluorescence (excitation 300 nm, emission 400 nm) [34]. For the mutase reaction, monitor the decrease in chorismate absorbance at 275 nm [34].
- Initial Rate Determination: Measure initial velocities at various substrate concentrations and fit to the Michaelis-Menten equation using nonlinear regression to extract k~cat~ and K~m~ values [34].
Isotopic Labeling and Computational Studies
The concerted nature of the [1,5]-sigmatropic shift in PchB was confirmed through deuterium labeling experiments coupled with NMR analysis, which demonstrated quantitative hydrogen transfer from C2 to C9 without scrambling [34]. This methodology provides unequivocal evidence for the pericyclic mechanism, distinguishing it from stepwise alternatives.
Computational approaches, particularly quantum mechanical/molecular mechanical (QM/MM) simulations, have become indispensable for studying enzymatic pericyclic reactions [34]. These methods:
- Model the electronic rearrangements during the concerted transition state at high levels of theory (e.g., DFT) [34]
- Analyze the evolution of molecular orbital symmetry throughout the reaction coordinate [34]
- Identify near attack conformations (NACs) that precede transition state formation [34]
- Calculate activation barriers and compare them with experimental kinetic data [34]
Table 3: Research Reagent Solutions for Pericyclic Reaction Studies
| Reagent/Material | Function in Research | Application Example |
|---|---|---|
| Deuterium-Labeled Substrates | Reaction mechanism tracing | Confirming [1,5]-sigmatropic shift in PchB via NMR [34] |
| Oxabicyclic Transition State Analogs | Enzyme structural studies | Determining X-ray structures of chorismate mutases [34] |
| Recombinant Enzymes | Kinetic and structural analysis | Site-directed mutagenesis to probe catalytic residues [34] |
| Quantum Mechanical/Molecular Mechanical (QM/MM) Software | Theoretical reaction modeling | Studying transition state geometry and electronic distribution [34] |
Research Workflow and Reaction Pathways
Diagram 1: Enzymatic Pericyclic Reaction Workflow
Diagram 2: PchB Catalyzed Reaction Pathways
Pericyclic reactions represent a cornerstone of modern organic chemistry, with principles of orbital symmetry conservation providing powerful predictive capabilities for reaction outcomes. The discovery of enzymatic counterparts to these reactions, particularly in biosynthetic pathways, has opened new frontiers in understanding biological catalysis. The PchB enzyme system exemplifies how nature exploits pericyclic mechanisms with remarkable efficiency and specificity, while also demonstrating the inherent promiscuity that may serve as an evolutionary starting point for new enzymatic functions.
Future research directions include elucidating the precise dynamic properties that enable enzymes to catalyze pericyclic reactions, engineering designer enzymes for specific pericyclic transformations, and exploring the full scope of pericyclic reactions in biochemical pathways. For drug development professionals, understanding these mechanisms provides valuable insights for designing enzyme inhibitors that mimic pericyclic transition states, potentially leading to novel therapeutic agents targeting essential biosynthetic pathways in pathogens and disease processes.
Mechanistic Tools and Pharmaceutical Applications in Drug Discovery
Within the broader thesis on reaction mechanisms in organic chemistry research, the elucidation of a reaction's pathway is a fundamental pursuit. Understanding whether a reaction occurs via a concerted mechanism or through a series of intermediates, and identifying the sequence of these elementary steps, is critical for predicting and optimizing outcomes in synthetic chemistry and drug development. This guide details two cornerstone experimental methodologies for mechanism elucidation: kinetic analysis and isotope labeling. Kinetic analysis provides quantitative data on reaction rates and their dependence on reactant concentrations, while isotope labeling allows researchers to track atomic movement and identify bond-breaking events. When used in concert, these techniques form a powerful toolkit for probing the inner workings of chemical transformations, enabling researchers to distinguish between plausible mechanisms and establish definitive reaction pathways.
Kinetic Analysis
Core Principles and Methodologies
Kinetic analysis involves measuring the rate of a chemical reaction and determining how this rate changes with variations in the concentrations of reactants, catalysts, and other species. The resulting rate law is a quantitative expression that provides deep insight into the mechanism's molecularity and the sequence of steps involved.
The reaction order with respect to each component is a key parameter determined experimentally. A zero-order dependence indicates the concentration of that species does not affect the rate, a first-order dependence shows a direct proportionality, and a second-order dependence suggests the rate is proportional to the square of its concentration. For catalytic reactions, determining the order in catalyst is essential for identifying the catalytically active species and the turnover-limiting step [35]. The order can be determined graphically or via the method of initial rates.
Advanced Kinetic Techniques
Reaction Progress Kinetic Analysis
Reaction Progress Kinetic Analysis (RPKA) is a powerful methodology for mechanistic studies of complex catalytic reactions. Unlike traditional initial rate methods, RPKA involves monitoring the reaction progress over a wide conversion range under synthetically relevant conditions. By using minimal experiments with varying initial concentrations, RPKA can deconvolute complex networks and provide information on catalyst activation, deactivation, and inhibition [35].
Variable Time Normalization Analysis
Variable Time Normalization Analysis (VTNA) is a graphical method to determine the order in catalyst. This technique simplifies the analysis by normalizing the reaction time, allowing for a direct visual assessment of the catalyst's order, which is crucial for understanding the catalytic cycle's kinetics [35].
Quantitative Kinetic Data and Analysis
Table 1: Common Reaction Orders and Their Mechanistic Implications
| Reaction Order | Measured Dependence | Key Mechanistic Implications |
|---|---|---|
| Zero Order | Rate is independent of reactant concentration | Saturation kinetics; often indicates a catalytic cycle limited by a step after substrate binding |
| First Order | Rate is directly proportional to concentration | Unimolecular step is rate-determining (e.g., dissociation, rearrangement) |
| Second Order | Rate is proportional to the square of concentration | Bimolecular step is rate-determining (e.g., oxidative addition, nucleophilic attack) |
| Fractional Order | Rate has a non-integer dependence on concentration | Complex mechanisms, often involving pre-equilibria or catalyst decomposition |
Table 2: Key Kinetic Isotope Effects (KIE) and Their Interpretation
| KIE Type | Isotopes Compared | Typical KIE (klight/kheavy) Value | Mechanistic Interpretation |
|---|---|---|---|
| Primary KIE | ¹H/²H, ¹²C/¹³C | 2 - 7 (for ¹H/²H) | Cleavage of a bond to the isotopically labeled atom occurs in the rate-determining step |
| Secondary KIE | ¹H/²H, ¹²C/¹³C | 1 - 1.5 (for ¹H/²H) | Change in hybridization or steric environment at the labeled atom in the rate-determining step |
| Inverse KIE (kheavy > klight) | ¹H/²H, ¹²C/¹³C | < 1 | A bond to the labeled atom is strengthened or a vibrational mode is stiffened in the transition state |
| Solvent KIE | ¹H/²H (H₂O/D₂O) | Variable | Proton transfer occurs in a step preceding the rate-determining step (pre-equilibrium) |
Experimental Protocols for Kinetic Studies
Protocol 1: Determining Reaction Order via the Method of Initial Rates
- Reaction Setup: Prepare a series of reaction vessels with varying initial concentrations of the reactant of interest, while keeping the concentrations of all other components constant.
- Initial Rate Measurement: For each reaction, monitor the concentration of a reactant or product over time, particularly in the very early stages (typically <5% conversion) where the rate is considered the initial rate. This can be done via techniques such as NMR spectroscopy, GC, or HPLC.
- Data Analysis: Plot the initial rate versus the initial concentration of the reactant. The resulting relationship indicates the order:
- A horizontal line suggests zero order.
- A linear plot suggests first order.
- A parabolic plot suggests second order.
- The slope of a log(rate) vs. log(concentration) plot gives the order directly.
Protocol 2: Reaction Progress Kinetic Analysis (RPKA) for Catalytic Reactions
- Experimental Design: Conduct two sets of experiments: one with different initial substrate concentrations (
[S]₀) and a constant catalyst concentration ([Cat]₀), and another with different[Cat]₀and a constant[S]₀. - In-Situ Monitoring: Use a technique like in-situ IR or ReactIR to monitor the concentration of a key species continuously throughout the entire reaction.
- Data Processing: Plot the reaction rate (d[S]/dt or d[P]/dt) against the concentration of the substrate
[S]or the catalyst[Cat]at different time points. The shapes of these "rate-concentration" curves are highly diagnostic for determining reaction orders and identifying complex behavior like catalyst inhibition or deactivation [35].
Isotope Labeling
Core Principles and Methodologies
Isotope labeling leverages atoms with distinct nuclear properties (e.g., ²H, ¹³C, ¹⁵N, ¹⁸O) to trace the fate of specific atoms or groups during a reaction. The fundamental principle is chemical equivalence: isotopes of an element exhibit nearly identical chemical behavior, but their physical properties (mass, radioactivity, nuclear spin) allow for detection [36]. This makes them powerful tracers for elucidating reaction mechanisms.
There are two primary categories of isotopes used:
- Radioactive Isotopes (e.g., ³H, ¹⁴C, ³²P): Tracked by their emitted radiation using scintillation counters or autoradiography. They are highly sensitive.
- Stable Isotopes (e.g., ²H, ¹³C, ¹⁵N, ¹⁸O): Analyzed by mass spectrometry (MS) for mass differences or nuclear magnetic resonance (NMR) for isotopic shifts in spectra [36].
Isotope Labeling Techniques and Applications
Kinetic Isotope Effects (KIE)
A KIE is observed when a reaction rate changes upon isotopic substitution. A primary KIE occurs when a bond to the isotopically labeled atom is broken or formed in the rate-determining step (RDS). A normal primary KIE (kH/kD > 1) confirms that the cleavage of that specific bond is a central feature of the RDS [35]. KIE studies can be conducted as intermolecular, intramolecular, or parallel experiments, each with specific advantages for minimizing experimental error.
Isotope Exchange and Incorporation
Isotope labeling can identify reversibility in reaction mechanisms. If a reactant undergoes H/D or ¹⁶O/¹⁸O exchange with the solvent before forming the final product, it indicates the existence of a reversible intermediate. For example, a novel enzymatic approach uses liver microsomal fractions to catalyze H/D and ¹⁶O/¹⁸O exchange into organic molecules, providing a mild and selective method for generating labeled compounds and probing exchangeable sites [37].
Site-Specific Labeling for Structural Biology
In biomolecular NMR, isotopic labeling is indispensable for studying the structure and dynamics of nucleic acids and proteins. Selective incorporation of ¹³C-methyl groups or aromatic ¹⁵N labels, combined with TROSY (Transverse Relaxation-Optimized Spectroscopy) experiments, allows researchers to push the size limit of biomolecules amenable to solution NMR, enabling the study of complexes exceeding 100 kDa [38].
Quantitative Isotope Data and Applications
Table 3: Common Isotopes and Their Applications in Mechanism Elucidation
| Isotope | Type | Key Detection Methods | Primary Applications in Mechanism Elucidation |
|---|---|---|---|
| Deuterium (²H) | Stable | ²H NMR, MS | Probing proton transfer steps; Kinetic Isotope Effects (KIE); tracing hydrogen migration (rearrangements) |
| Carbon-13 (¹³C) | Stable | ¹³C NMR, MS | Tracing carbon skeletons in rearrangement reactions; determining metabolic pathways (MFA) |
| Nitrogen-15 (¹⁵N) | Stable | ¹⁵N NMR, MS | Studying reaction mechanisms involving amines, nitriles, and other N-containing functional groups |
| Oxygen-18 (¹⁸O) | Stable | MS | Tracing the origin of oxygen atoms in oxidation reactions; distinguishing between different mechanistic pathways for oxygen incorporation |
| Carbon-14 (¹⁴C) | Radioactive | Scintillation Counting | Ultra-sensitive tracing of carbon atoms in complex matrices or low-concentration reactions |
Table 4: Research Reagent Solutions for Isotope Labeling
| Research Reagent / Material | Function / Application | Brief Explanation |
|---|---|---|
| D₂O (Deuterated Water) | Heavy solvent for H/D exchange | Source of deuterium for labeling labile hydrogens; used to probe reversible steps and reaction mechanisms. |
| H₂¹⁸O (Oxygen-18 Water) | Heavy solvent for ¹⁶O/¹⁸O exchange | Source of ¹⁸O for labeling oxygen-containing functional groups (e.g., carboxylic acids, aldehydes). |
| ¹³C-labeled Substrates (e.g., ¹³C-glucose) | Metabolic tracer | Used in Metabolic Flux Analysis (MFA) to map carbon flow through biochemical pathways in living systems. |
| SILAC Media (Stable Isotope Labeling with Amino acids in Cell culture) | Quantitative proteomics | Incorporates "heavy" ¹³C/¹⁵N-lysine/arginine into proteins for accurate quantification by mass spectrometry. |
| Methyltransferases (e.g., Msp, 2Bst) | Chemo-enzymatic labeling | Enzymes that install ¹³C-methyl groups at specific sites in DNA/RNA for NMR studies of large biomolecular complexes [38]. |
| Liver Microsomal Fractions | Enzymatic isotope exchange | A cost-effective catalytic source containing enzymes (e.g., CYPs) that can catalyze H/D and ¹⁶O/¹⁸O exchange into small molecules [37]. |
Experimental Protocols for Isotope Labeling
Protocol 3: Conducting an Intermolecular Kinetic Isotope Effect (KIE) Study
- Parallel Reactions: Set up two separate but otherwise identical reactions: one with the natural abundance (light) substrate and another with the isotopically labeled (heavy) substrate (e.g., C-H vs. C-D bond).
- Rate Measurement: Accurately measure the initial reaction rate for each reaction (
k_lightandk_heavy) using a suitable analytical method (e.g., GC, HPLC, NMR). - KIE Calculation: Calculate the KIE as the ratio
k_light / k_heavy. A value significantly greater than 1 (typically 2-7 for ¹H/²H) indicates that cleavage of the bond to the labeled atom is occurring in the rate-determining step.
Protocol 4: Enzymatic H/D Exchange Using Liver Microsomal Fractions
- Preparation: Incubate the target compound (e.g., progesterone) with a pooled rat liver microsomal (RLM) fraction in a deuterated buffer (PBS in D₂O) [37].
- Control Experiment: Run a parallel control incubation in protiated (H₂O) buffer under identical conditions.
- Analysis: After a set period, quench the reaction and analyze the products using LC-HRMS. The number and position of deuterium incorporations are identified by the mass shift and compared to the control. This pattern can reveal sites susceptible to enzymatic exchange, often correlating with known metabolic soft spots [37].
Integrated Workflows and Emerging Technologies
Modern mechanism elucidation increasingly relies on integrating multiple techniques and leveraging new technologies to accelerate discovery.
Complementary Use of Techniques
Kinetic analysis and isotope labeling are rarely used in isolation. For example, a Hammett study (which correlates reaction rate with substituent constants) might suggest a change in mechanism for electron-donating versus electron-withdrawing groups. This hypothesis can be tested decisively with a KIE experiment; a change in the magnitude of the KIE across the series would provide strong supporting evidence for the proposed mechanistic shift [35]. Similarly, identifying a potential intermediate through kinetic analysis (e.g., a burst phase) can be followed by isotope labeling experiments to trap or characterize that intermediate.
High-Throughput and Automated Experimentation
The traditional one-reaction-at-a-time approach to kinetic and labeling studies is being transformed by High-Throughput Experimentation (HTE). Automated platforms, particularly those using flow chemistry, enable the rapid screening of vast reaction parameter spaces (e.g., catalyst, solvent, temperature, concentration) [39] [40]. Flow chemistry is especially useful for kinetic studies due to its precise control over reaction time and temperature, and for handling hazardous reagents generated in situ for isotope labeling [39]. These automated systems can be coupled with inline analytical techniques, such as IR or MS, to provide real-time data for reaction monitoring and optimization.
Computational Integration
Density Functional Theory (DFT) and other computational methods have become indispensable partners to experimental mechanism elucidation. Calculations can predict transition state energies, geometries, and isotopic properties, which can be directly compared to experimental kinetic and KIE data [35]. This synergy between computation and experiment allows for the proposal and validation of mechanically detailed reaction pathways with high confidence.
Workflow Diagrams
Mechanism Elucidation Workflow
Modern Reaction Optimization
Spectroscopic Methods (NMR, IR) and Computational Chemistry for Studying Transition States
Precise understanding of reaction kinetics, requiring accurate transition states and energy barriers, is central to organic synthesis and drug development [41]. A transition state represents a molecular configuration at a local energy maximum along a reaction pathway—a fleeting, non-isolable structure with partial bonds and an extremely short lifetime measured in femtoseconds [42]. Unlike reactive intermediates, which exist at energy minima and can sometimes be observed directly, transition states represent the "point of no return" in chemical reactions, making their characterization one of the most challenging problems in physical organic chemistry [42]. This whitepaper examines the integrated application of spectroscopic methods and computational chemistry to overcome this challenge, providing researchers with practical methodologies for transition state analysis within broader reaction mechanism studies.
Theoretical Foundation of Transition States
Fundamental Characteristics
A transition state is mathematically defined as a geometry with a zero first derivative of energy with respect to all nuclear coordinates, and a second derivative that is positive for all but one geometric coordinate, which has negative curvature [43]. In practical terms, this represents a saddle point on the potential energy surface—a maximum along the reaction pathway but a minimum in all other directions.
Key properties distinguish transition states from other species:
- Partial bonds exist in the process of forming or breaking, represented as dashed lines in structural drawings [44]
- Extremely short lifetimes on the order of femtoseconds, making direct experimental observation impossible [42]
- Geometry intermediate between reactants and products, though not necessarily perfectly symmetrical [45]
- Highest energy point along the reaction pathway for an elementary step, determining the reaction's activation energy [42]
The Concept of Virtual Transition States
For complex reactions involving multiple pathways in parallel or consecutive steps, the concept of "virtual transition states" has recently emerged. This describes a weighted average of several contributing real transition states lying close in energy. The virtual transition state simplifies the treatment of complex kinetic phenomena such as Hammett plot interpretations and kinetic isotope effects, particularly relevant for enzymatic reactions in pharmaceutical research [46].
Computational Chemistry Approaches
Traditional Quantum Chemistry Methods
Density Functional Theory (DFT) remains the mainstream quantum chemical method for transition state searches, despite its inherent trade-offs between accuracy and computational cost [41]. Traditional computational protocols involve:
Geometry Optimization Algorithms: Quasi-Newton methods that compute the Hessian matrix (second derivatives of energy with respect to nuclear motion) and move nuclei to increase energy along the reaction coordinate while decreasing energy in other directions [43].
Reaction Coordinate Techniques:
- Linear Synchronous Transit (LST): Assumes atoms move linearly from reactant to product positions
- Quadratic Synchronous Transit (QST): Assumes atoms follow a parabolic path between endpoints
- Intrinsic Reaction Coordinate (IRC): Follows the steepest descent path from the transition state to reactants and products [43]
Level of Theory Considerations: Transition structures are generally more difficult to describe accurately than equilibrium geometries. Lower levels of theory such as semiempirical methods, local density approximation DFT, and ab initio methods with small basis sets often perform poorly for transition state prediction [43].
Table 1: Performance Comparison of Computational Methods for Transition State Optimization
| Computational Method | Typical Success Rate | Computational Cost | Key Applications |
|---|---|---|---|
| B3LYP/def2-SVP | Variable (lower for fluorinated systems) | Moderate | Preliminary screening |
| ωB97X/pcseg-1 | Higher for challenging systems | High | Accurate barrier prediction |
| M08-HX/pcseg-1 | Higher for challenging systems | High | Complex reaction landscapes |
| Machine Learning (React-OT) | ~90% | Very Low (0.4 seconds) | High-throughput screening |
Machine Learning Revolution
Recent advances in machine learning have dramatically accelerated transition state prediction, with models like React-OT generating accurate structures in less than 0.4 seconds—nearly four orders of magnitude faster than rigorous DFT computations [41] [47].
Architecture and Training: The React-OT model uses optimal transport theory, starting from an estimate generated by linear interpolation that positions each atom halfway between its reactant and product positions in three-dimensional space. This provides a superior initial guess compared to random starting points [47].
Performance Metrics: Modern ML potentials like DeePEST-OS achieve remarkable accuracy, exhibiting root mean square deviations of 0.12 Å for transition state geometries and mean absolute errors of 0.60 kcal/mol for reaction barriers across external test reactions [41].
Broad Elemental Coverage: Generic ML potentials now cover up to ten element types, dramatically extending from traditional four-element coverage and enabling applications to pharmaceuticals containing halogens, sulfur, and phosphorus [41].
Diagram 1: Machine Learning Workflow for Transition State Prediction. This streamlined process enables high-throughput screening of reaction barriers.
Spectroscopic Methods for Indirect Characterization
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR transitions are slow compared to most chemical transformations or conformational equilibria, meaning conventional NMR cannot directly detect transition states [48]. However, advanced NMR techniques provide crucial indirect information:
Dynamic NMR (DNMR): By exploring spectral changes as a function of temperature, DNMR accesses information about transformation rates and activation energies for processes that are slow on the NMR timescale [48]. For example, in the study of 4-formylaminoantipyrine, two distinct species were observed due to slow rotation around the carbon-nitrogen partial double bond at room temperature [48].
Computational NMR Analysis: DFT calculations of NMR parameters (chemical shifts and coupling constants) provide powerful validation for proposed structures and reaction pathways [49]. The best computational approaches combine GIAO (gauge-including atomic orbital) or CSGT (continuous set of gauge transformation) methods with appropriate functionals and basis sets [49].
Infrared (IR) Spectroscopy
While limited for direct transition state observation, IR spectroscopy plays a critical role in structural validation:
Vibrational Frequency Calculations: Comparing DFT-calculated IR spectra for putative transition states with experimental data provides validation, as demonstrated in the structural elucidation of arsenicin A, where this approach was necessary due to inconclusive NMR data [49].
Experimental Validation: For polyarsenical compounds like arsenicin A, comparison of experimental and calculated IR spectra provided the definitive structural assignment when NMR data proved insufficient [49].
Integrated Workflows and Experimental Protocols
Combined Computational-Spectroscopic Protocol
Phase 1: Initial Computational Screening
- Structure Preparation: Generate 3D structures of reactants and products using chemical sketching tools or database retrieval
- Conformational Search: Perform molecular mechanics or semi-empirical conformational sampling to identify low-energy conformers
- Machine Learning TS Prediction: Apply React-OT or similar ML models to generate initial transition state guesses in seconds [47]
- Quantum Chemical Refinement: Optimize promising candidates using DFT methods (ωB97X or M08-HX functionals with polarized basis sets recommended) [50]
Phase 2: Spectroscopic Validation
- NMR Chemical Shift Calculation: Compute ( ^1H ) and ( ^{13}C ) chemical shifts for proposed intermediates and products using GIAO-DFT protocols [49]
- Comparison with Experimental Data: Validate computational models by comparing calculated NMR parameters with experimental spectra
- IR Frequency Calculation: Compute vibrational frequencies for proposed structures to compare with experimental IR data when available [49]
Phase 3: Kinetic Analysis
- Activation Energy Calculation: Determine energy barriers from transition state energies
- Isotope Effect Prediction: Calculate kinetic isotope effects for comparison with experimental data
- Rate Constant Prediction: Apply transition state theory to predict rate constants for comparison with kinetic measurements
Table 2: Essential Research Reagent Solutions for Transition State Analysis
| Research Tool | Function/Application | Key Features |
|---|---|---|
| DFT Software (Gaussian, ORCA) | Quantum chemical optimization of transition states | Supports various functionals (ωB97X, M08-HX); IRC calculations |
| Machine Learning Potentials (DeePEST-OS, React-OT) | Rapid transition state initial guess generation | Near-instant prediction; broad elemental coverage |
| DNMR Spectroscopy System | Study of slow conformational exchanges | Variable temperature capability; high field magnet |
| GIAO-DFT Protocol | Calculation of NMR parameters from optimized structures | Predicts chemical shifts and coupling constants |
| IR Computational Modules | Calculation of vibrational frequencies | Validation against experimental spectra |
Protocol for Challenging Systems: HFC/HFE Atmospheric Degradation
For particularly challenging systems like hydrofluorocarbon (HFC) and hydrofluoroether (HFE) reactions with hydroxyl radicals, specialized protocols have been developed:
Bitmap Representation and CNN Analysis: Convert 3D molecular geometry into 2D bitmaps, then apply convolutional neural networks (ResNet50 architecture) with genetic algorithms to assess initial guess quality [50]. This approach achieved success rates of 81.8% for HFCs and 80.9% for HFEs where traditional methods often fail [50].
Multi-level Theoretical Validation: Combine semi-empirical methods for initial screening with higher-level DFT (ωB97X/pcseg-1) for final energy evaluations [50].
Applications in Pharmaceutical Research
The integration of computational and spectroscopic methods for transition state analysis has proven particularly valuable in pharmaceutical research:
Retrosynthesis Planning: ML potentials with broad elemental coverage enable retrosynthesis of complex pharmaceuticals containing heteroatoms, such as Zatosetron, previously unachievable with earlier methods [41].
Enzyme Mechanism Elucidation: The virtual transition state concept simplifies interpretation of kinetic isotope effects in enzymatic reactions, facilitating drug design targeting enzyme active sites [46].
Atmospheric Lifetime Prediction: For pharmaceutical propellants and inhalers, transition state modeling predicts atmospheric degradation rates and global warming potential, informing environmentally conscious design [50].
Diagram 2: Integrated Workflow for Pharmaceutical Application. This iterative process refines mechanistic understanding and enables rational drug design.
The synergistic combination of spectroscopic methods and computational chemistry has transformed transition state analysis from an insurmountable challenge to a tractable problem in reaction mechanism research. While NMR and IR spectroscopy provide essential experimental validation for proposed structures and pathways, advanced computational methods—particularly machine learning potentials—have dramatically accelerated the discovery process. For drug development professionals, these integrated approaches enable rational design of synthetic routes, elucidation of enzymatic mechanisms, and prediction of environmental fate for pharmaceutical compounds. As machine learning algorithms continue to evolve and spectroscopic techniques advance, the precision and efficiency of transition state analysis will further accelerate, solidifying its role as a cornerstone of modern organic chemistry research.
Cross-Coupling Reactions (Suzuki-Miyaura, Buchwald-Hartwig) in API Synthesis
Cross-coupling reactions represent one of the most significant advancements in organic chemistry over the past half-century, permanently transforming synthetic strategies for constructing carbon-carbon (C–C) and carbon-heteroatom (C–X) bonds [51]. These metal-catalyzed transformations enable the precise connection of molecular fragments under mild conditions with exceptional functional group tolerance. Within pharmaceutical development, cross-coupling reactions have become indispensable tools for constructing active pharmaceutical ingredients (APIs) and drug candidates, particularly those containing biaryl, heteroaromatic, and aromatic amine motifs prevalent in modern therapeutics [51] [52]. The profound impact of these methods was recognized with the 2010 Nobel Prize in Chemistry, awarded for palladium-catalyzed cross-couplings [51].
This technical guide focuses on two cornerstone cross-coupling methodologies with exceptional utility in API synthesis: the Suzuki-Miyaura coupling for C–C bond formation and the Buchwald-Hartwig amination for C–N bond formation. When framed within the broader context of reaction mechanism research in organic chemistry, these transformations exemplify how deep mechanistic understanding enables the rational design of synthetic methodologies that address complex challenges in drug development [53] [54]. The convergence of mechanistic studies, catalyst design, and increasingly sophisticated optimization approaches continues to expand the applications of these pivotal reactions in pharmaceutical synthesis.
Fundamental Reaction Mechanisms
Unified Catalytic Cycle for Cross-Coupling Reactions
Palladium-catalyzed cross-coupling reactions follow a general mechanistic pathway involving three fundamental steps: oxidative addition, transmetalation, and reductive elimination [55]. The catalytic cycle begins with a palladium(0) species, which undergoes oxidative addition with an organic electrophile (typically an aryl or vinyl halide), forming a palladium(II) complex. Subsequently, transmetalation occurs where an organometallic nucleophile transfers its organic group to the palladium center. Finally, reductive elimination produces the coupled product while regenerating the active palladium(0) catalyst [51] [55]. This mechanistic framework provides the foundation for both Suzuki-Miyaura and Buchwald-Hartwig reactions, with variations occurring primarily in the transmetalation step.
Suzuki-Miyaura Cross-Coupling Mechanism
The Suzuki-Miyaura reaction specifically couples organoboron nucleophiles with organic electrophiles to form C–C bonds [56]. Unlike other cross-couplings, the Suzuki mechanism requires activation of the boron atom via a base [56]. This base activation enhances the polarization of the organic ligand on boron, facilitating the transmetalation step where the organic group transfers from boron to palladium [56]. The exceptional stability, low toxicity, and commercial availability of organoboron reagents, combined with their tolerance toward aqueous conditions and various functional groups, have established Suzuki-Miyaura coupling as one of the most widely applied C–C bond-forming methods in API synthesis [56] [52] [55].
Buchwald-Hartwig Amination Mechanism
The Buchwald-Hartwig amination, developed in 1994, enables the formation of C–N bonds between aryl halides and amines [51]. This transformation proceeds through a similar palladium catalytic cycle but differs in the transmetalation step. Instead of organometallic transmetalation, the amine nucleophile first deprotonates in the presence of base, then coordinates to the palladium center. Reductive elimination subsequently forms the C–N bond, yielding aryl amine products [57]. This methodology has revolutionized the synthesis of aromatic amines, privileged structural motifs in numerous pharmaceuticals and agrochemicals.
Recent Methodological Advances and Applications in API Synthesis
Suzuki-Miyaura Coupling: Expanded Substrate Scope and Greener Protocols
Recent advances in Suzuki-Miyaura coupling have dramatically expanded its applications in API synthesis through developments in catalyst design, substrate scope, and reaction conditions [58] [52]. Modern research focuses on increasing the reactivity of challenging substrates such as aryl chlorides (significantly less expensive but less reactive than bromides or iodides), enabling alkyl-alkyl couplings, and developing sustainable reaction media [56] [52].
Catalyst Development: Significant progress has been made with palladium catalysts incorporating N-heterocyclic carbene (NHC) ligands, which enhance stability and activity toward sterically hindered and electronically deactivated substrates [56]. Phosphine-free palladium systems have also emerged as economical and efficient alternatives [56]. For example, Pd/P,O-ligand complexes and palladium/diazabutadiene systems demonstrate excellent activity across diverse substrate classes [56].
Sustainable Reaction Media: Recent methodologies have enabled Suzuki couplings in aqueous solvents, with micellar conditions proving particularly effective for hydrophobic pharmaceutical intermediates [56]. Aqueous Suzuki-Miyaura coupling with ultralow palladium loading and straightforward product separation represents significant progress toward environmentally benign API synthesis [56].
API Synthesis Applications: A notable application includes the synthesis of Lumacaftor, a cystic fibrosis medication, using a Pd-catalyzed C(sp²)-C(sp³) Suzuki coupling with specifically designed monophosphine ligands [56]. The ability to couple unactivated secondary alkyl halides at room temperature further demonstrates the method's advancing capabilities for constructing complex pharmaceutical architectures [56].
Buchwald-Hartwig Amination: Enabling Targeted Protein Degradation Platforms
Recent applications of Buchwald-Hartwig amination in pharmaceutical development highlight its critical role in emerging therapeutic modalities. A 2024 study demonstrated its application in synthesizing cereblon (CRBN) binders for targeted protein degradation (TPD), a pioneering therapeutic strategy [57]. Researchers developed optimized Buchwald-Hartwig conditions for the direct cross-coupling of unprotected glutarimides with amines, streamlining the synthesis of alternative CRBN binders beyond traditional immunomodulatory drugs [57]. This methodology enabled the rapid construction of a 30-compound library of potential degraders, showcasing the power of modern C–N coupling in generating diverse scaffolds for drug discovery [57].
Table 1: Key Advances in Cross-Coupling Reactions for API Synthesis
| Reaction Type | Recent Advancement | Application in API Synthesis | Key Benefit |
|---|---|---|---|
| Suzuki-Miyaura | Room-temperature coupling of alkyl bromides [56] | Synthesis of complex alkyl-aryl scaffolds | Mild conditions compatible with base-sensitive functionalities |
| Suzuki-Miyaura | Aqueous micellar conditions [56] | Coupling of hydrophobic intermediates | Reduced organic solvent waste; simplified workup |
| Suzuki-Miyaura | C(sp²)-C(sp³) coupling with designed ligands [56] | Lumacaftor API synthesis | Improved selectivity for challenging bond formations |
| Buchwald-Hartwig | Direct coupling of unprotected glutarimides [57] | Cereblon binder library for targeted protein degradation | Streamlined synthesis of degraders with varying properties |
Experimental Protocols and Optimization Strategies
General Suzuki-Miyaura Coupling Procedure
A typical experimental protocol for Suzuki-Miyaura coupling involves the following steps [56] [52]:
Reaction Setup: In an inert atmosphere glove box or using Schlenk techniques, add the aryl halide (1.0 equiv), organoboron reagent (1.2-1.5 equiv), and base (2.0-3.0 equiv) to a reaction vessel containing a stir bar.
Catalyst Addition: Add the palladium catalyst (0.5-5 mol%) and appropriate ligand (if required; 1-10 mol%) to the mixture. Common catalyst systems include:
- Pd(OAc)₂ with tri-tert-butylphosphine for aryl chlorides
- Pd₂(dba)₃ with tricyclohexylphosphine for aryl triflates
- Pd(PPh₃)₄ for activated aryl bromides
Solvent Introduction: Add degassed solvent (e.g., toluene/water mixtures, DMF, dioxane, or aqueous micellar systems) via syringe.
Reaction Execution: Heat the reaction mixture at the appropriate temperature (room temperature to 100°C) with vigorous stirring for the specified time (typically 2-24 hours).
Workup and Purification: After completion (monitored by TLC or LCMS), cool the reaction to room temperature, dilute with water and ethyl acetate, and separate the layers. Wash the organic layer with brine, dry over anhydrous Na₂SO₄, filter, and concentrate under reduced pressure. Purify the crude product via flash column chromatography or recrystallization.
Optimized Buchwald-Hartwig Protocol for Cereblon Binder Synthesis
A recently developed optimized protocol for Buchwald-Hartwig amination in CRBN binder synthesis exemplifies modern application [57]:
Reaction Setup: Charge a microwave vial with aryl halide (1.0 equiv), amine nucleophile (1.2 equiv), Pd₂(dba)₃ (2-5 mol%), BrettPhos or RuPhos type ligand (4-10 mol%), and sodium tert-butoxide (1.5-2.0 equiv).
Solvent Addition: Add degassed 1,4-dioxane or toluene to the mixture via syringe.
Reaction Conditions: Heat the reaction at 80-100°C for 12-16 hours with stirring.
Purification: After completion, cool the reaction mixture, dilute with ethyl acetate, filter through a pad of Celite, and concentrate under reduced pressure. Purify the residue via preparative HPLC or flash chromatography.
Critical Reaction Parameters and Optimization Guidance
Successful cross-coupling implementation requires careful optimization of several key parameters:
Base Selection: The choice of base significantly impacts Suzuki-Miyaura reactions. Carbonate bases (K₂CO₃, Cs₂CO₃) are commonly employed, while powdered KF can activate boron with minimal impact on base-labile functional groups [56]. For Buchwald-Hartwig aminations, strong bases like NaO-t-Bu are typically required [57].
Solvent Systems: Common solvents include toluene, dioxane, DMF, and their aqueous mixtures. Recent advances demonstrate the effectiveness of aqueous micellar systems for Suzuki couplings and neat conditions for specific applications [56].
Catalyst-Ligand Systems: Ligand selection profoundly influences catalyst activity and stability. Bulky, electron-rich phosphines facilitate oxidative addition of less reactive electrophiles, while specialized ligands enable challenging C(sp³)-C(sp²) couplings [56] [52].
Table 2: Optimization Guide for Cross-Coupling Reaction Parameters
| Parameter | Suzuki-Miyaura Options | Buchwald-Hartwig Options | Impact on Reaction |
|---|---|---|---|
| Catalyst | Pd(OAc)₂, Pd₂(dba)₃, Pd(PPh₃)₄ | Pd₂(dba)₃, Pd(OAc)₂ | Influences initial activation and turnover frequency |
| Ligand | P(t-Bu)₃, SPhos, XPhos | BrettPhos, RuPhos, XantPhos | Determines substrate scope and functional group tolerance |
| Base | K₂CO₃, Cs₂CO₃, K₃PO₄ | NaO-t-Bu, Cs₂CO₃, K₃PO₄ | Activates nucleophile; critical for transmetalation |
| Solvent | Toluene/H₂O, DMF, dioxane, aqueous micellar | Toluene, dioxane, DMF | Affects solubility, catalyst stability, and reaction temperature |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagent Solutions for Cross-Coupling Reactions
| Reagent Category | Specific Examples | Function in Reaction |
|---|---|---|
| Palladium Sources | Pd(OAc)₂, Pd₂(dba)₃, Pd(PPh₃)₄ | Catalytic precursor; source of active Pd(0) species |
| Phosphine Ligands | P(t-Bu)₃, XPhos, SPhos, BrettPhos | Stabilize Pd center; facilitate oxidative addition; prevent aggregation |
| Organoboron Reagents | Arylboronic acids, alkylboronic acids, potassium trifluoroborates, boronic esters | Nucleophilic coupling partner in Suzuki reactions |
| Bases | K₂CO₃, Cs₂CO₃, NaO-t-Bu, K₃PO₄ | Activate boron reagent (Suzuki) or deprotonate amine (Buchwald-Hartwig) |
| Solvents | Degassed toluene, 1,4-dioxane, DMF, aqueous buffers | Reaction medium; must be oxygen-free to maintain catalyst activity |
Emerging Trends: Data-Driven Reaction Optimization
The field of cross-coupling is increasingly embracing data science and machine learning to accelerate reaction optimization and prediction [53] [59] [54]. Recent studies demonstrate the development of quantitative structure-reactivity models that predict outcomes for palladium-catalyzed cross-couplings based on molecular descriptors of the substrates [53]. These approaches combine high-throughput experimentation (HTE) with machine learning algorithms to navigate complex reaction parameter spaces efficiently [59] [54].
Closed-loop systems that autonomously design, execute, and analyze experiments using machine learning optimization algorithms represent the cutting edge of reaction optimization methodology [59]. However, the most successful approaches maintain a synergy between artificial intelligence and human chemical intuition, leveraging the rapid exploration capabilities of AI while benefiting from the deep mechanistic understanding of experienced chemists [59]. These technologies are particularly valuable in pharmaceutical development, where they can significantly reduce optimization time for key bond-forming steps in API synthesis.
Suzuki-Miyaura and Buchwald-Hartwig cross-coupling reactions have matured into indispensable tools for constructing C–C and C–N bonds in API synthesis. Through continuous mechanistic investigation and catalyst design, these methodologies now address increasingly challenging synthetic problems, from constructing sterically hindered biaryl systems to enabling emerging therapeutic modalities like targeted protein degradation. The ongoing integration of data science and automation with fundamental chemical principles promises to further accelerate the deployment of these transformative reactions in pharmaceutical development, solidifying their pivotal role in drug discovery and development for the foreseeable future.
This technical guide provides an in-depth examination of the reaction mechanisms and synthetic pathways for two pivotal therapeutic agents: losartan, an angiotensin II receptor blocker (ARB) for hypertension, and abemaciclib, a cyclin-dependent kinase (CDK) 4/6 inhibitor for breast cancer. Framed within a broader thesis on organic reaction mechanisms, this analysis highlights how strategic bond formation and catalyst selection in process chemistry directly influence drug efficacy, safety profiles, and manufacturing sustainability. The comparative study of these molecules—one a cardiovascular staple and the other a modern oncology treatment—showcases the evolution of synthetic philosophy in pharmaceutical development, where green chemistry principles and mechanistic toxicology are increasingly integrated from discovery through to scale-up [60] [61] [62].
Losartan: Synthetic Pathways and Green Chemistry Innovations
Drug Profile and Significance
Losartan, the first approved angiotensin II receptor blocker, is a mainstay in treating hypertension and diabetic nephropathy. Its mechanism involves selective antagonism of the angiotensin II type 1 (AT₁) receptor, leading to vasodilation and reduced aldosterone secretion [60]. Structurally, it features a biphenyl scaffold tethered to a tetrazole ring and a chloroimidazole group, motifs that present distinct synthetic challenges [63].
Conventional Synthetic Routes
Traditional losartan synthesis has relied on multiple strategic approaches, each with specific mechanistic considerations:
- Ullmann Biaryl Coupling: Early routes employed copper-promoted coupling of 2-iodobenzonitrile and 4-iodotoluene at high temperatures (180–190°C). This method suffered from low yields and required cumbersome purification via column chromatography and Kugelrohr distillation to isolate the key biphenyl intermediate, 4′-methyl-[1,1′-biphenyl]-2-carbonitrile [63].
- Meyers Oxazolidine Route: This method involves a multi-step sequence starting from 2-methoxybenzoyl chloride, proceeding through an oxazolidine intermediate formed with 2-amino-2-methyl-1-propanol. The key step is a displacement reaction with (4-methylphenyl)magnesium bromide, followed by ring fragmentation to install the nitrile group [63].
- Sequential Alkylation-Cyclization: A common industrial pathway involves alkylation of (2-butyl-4-chloro-1H-imidazol-5-yl)methanol with 4′-(bromomethyl)-[1,1′-biphenyl]-2-carbonitrile. The critical tetrazole ring formation then employs trimethyltin azide—a highly toxic reagent requiring careful workup to remove tin contaminants—or direct cyclization with sodium azide in the presence of an ammonium salt [63].
Table 1: Key Intermediates in Traditional Losartan Synthesis
| Intermediate Name | Chemical Structure | Role in Synthesis | Synthetic Challenge |
|---|---|---|---|
| 4′-(bromomethyl)-[1,1′-biphenyl]-2-carbonitrile | Biphenyl core with bromomethyl and nitrile groups | Serves as electrophile for imidazole alkylation; nitrile is precursor to tetrazole | Benzylic bromide instability; requires careful handling and purification |
| (2-butyl-4-chloro-1H-imidazol-5-yl)methanol | Chloroimidazole with hydroxymethyl group | Nucleophile for alkylation; provides imidazole scaffold | Synthesis requires controlled chlorination conditions |
| Trimethyltin tetrazole adduct | Tetrazole group coordinated to tin | Tetrazole protection intermediate | High toxicity of organotin compounds; difficult removal |
Advanced Green Synthesis Using Sustainable Catalysis
Recent innovations focus on addressing the limitations of traditional methods, particularly the use of toxic reagents and problematic solvents linked to nitrosamine impurities like N-nitrosodimethylamine (NMDA) that prompted drug recalls [61].
A 2025 study demonstrates a sustainable synthetic route using palladium nanoparticles (PdNPs) derived from the brown seaweed Sargassum incisifolium. These bio-sourced catalysts offer an eco-friendly alternative for the pivotal Suzuki–Miyaura cross-coupling reaction [61].
Experimental Protocol: Green Suzuki–Miyaura Coupling
- Reaction Setup: A mixture of 2-bromobenzonitrile (1.0 equiv) and 4-methylphenylboronic acid (1.2 equiv) in aqueous acetone.
- Catalyst System: Seaweed-derived PdNPs (0.5 mol% Pd), K₂CO₃ as base.
- Reaction Conditions: Mild conditions (70°C for 2 hours) under ambient atmosphere.
- Workup: Simple filtration to recover the catalyst, followed by solvent evaporation and purification.
- Key Result: The reaction achieves a 98% yield of the biphenyl intermediate 4′-methyl-biphenyl-2-carbonitrile, which can be subsequently brominated using N-bromosuccinimide (NBS) under LED light to advance the synthesis [61].
Mechanistic Advantages: The seaweed extract contains polyphenols that reduce Pd(II) to Pd(0) and polysaccharides that stabilize the nanoparticles, preventing aggregation. The catalyst exhibits excellent recyclability over multiple runs without significant loss of activity, and the aqueous acetone solvent system eliminates the need for toxic dipolar aprotic solvents like DMF [61].
Table 2: Comparison of Losartan Synthetic Methods
| Synthetic Parameter | Traditional Ullmann Route | Suzuki–Miyaura with PdNPs |
|---|---|---|
| Key Coupling Reaction | Copper-mediated coupling | Palladium-catalyzed cross-coupling |
| Reaction Temperature | 180–190°C | 70°C |
| Overall Yield | Not specified in sources | 27% (prioritizing green metrics over yield) |
| Catalyst System | Copper powder | Bio-derived Pd nanoparticles (8–10 nm) |
| Green Chemistry Merit | Poor atom economy, high energy | Renewable catalyst, aqueous solvent, recyclable |
Figure 1: Sustainable Losartan Synthesis Workflow. This flowchart illustrates the key steps in the green synthetic route for losartan, highlighting the pivotal Suzuki–Miyaura coupling using bio-derived PdNPs and subsequent functionalization steps.
Abemaciclib: Mechanisms of Action and Cutaneous Toxicity
Drug Profile and Therapeutic Significance
Abemaciclib is an oral inhibitor of cyclin-dependent kinases 4 and 6 (CDK4/6), approved for HR+/HER2- breast cancer in both metastatic and early-stage, high-risk settings [64] [65]. By inhibiting the CDK4/6 kinase activity, abemaciclib prevents phosphorylation of the retinoblastoma protein, thereby inducing G1 cell cycle arrest and suppressing tumor proliferation [65].
Clinical Adverse Reaction Profile
The safety profile of abemaciclib is characterized by specific, manageable adverse reactions. Real-world evidence from a 2025 retrospective study of 216 patients identified the most common adverse effects (AEs) and their risk factors [66]:
- Diarrhea: Occurred in 81-90% of patients across clinical trials, with 8-20% experiencing Grade 3 severity. The median time to onset was 6-8 days, typically managed with antidiarrheal agents like loperamide, dose adjustments, and increased fluid intake [64] [65].
- Hematologic Toxicity: Neutropenia occurred in 37-46% of patients, with Grade ≥3 events in 19-32%. Neutropenia typically manifests within the first treatment month (median time to first episode: 29 days) [64].
Table 3: Risk Factors for Common Abemaciclib Adverse Reactions
| Adverse Reaction | Risk Factors | Protective Factors | Clinical Management |
|---|---|---|---|
| Grade 2-3 Diarrhea | Age stratification, pre-existing gastrointestinal diseases | Not specified | Early antidiarrheal therapy (loperamide), dose interruption/reduction, fluid hydration |
| Grade 3-4 Neutropenia | Higher ECOG performance status | Higher baseline BMI, higher baseline WBC count, higher baseline albumin | Regular blood monitoring (every 2 weeks for first 2 months), dose modification, growth factor support |
Molecular Mechanisms of Cutaneous Toxicity
A 2025 mechanistic study investigated abemaciclib-induced skin reactions at the cellular level, revealing an oxidative inflammation pathway as the primary driver of cytotoxicity in human keratinocytes (HaCaT cells) [62].
Experimental Protocol: Keratinocyte Cytotoxicity Assessment
- Cell Model: Human keratinocyte (HaCaT) cell line.
- Treatment Conditions: Exposure to abemaciclib (0.1-10 µM) for 24 hours.
- Assays Performed: Cytotoxicity (IC₅₀ determination), apoptosis/necrosis measurement, oxidative stress markers, inflammatory cytokine secretion.
- Key Findings: The half-maximal inhibitory concentration (IC₅₀) was ≥24.18 µM. Notably, the maximum pro-inflammatory effects were observed at the lowest concentration tested (0.1 µM), demonstrating a non-linear dose-response relationship [62].
Mechanistic Insights: Abemaciclib induced significant oxidative damage and apoptosis even at low concentrations (0.1 µM). There was a marked increase in secretion of pro-inflammatory cytokines MCP-1, IL-6, and IL-8 at 0.1 µM, while higher concentrations (1-10 µM) showed diminished inflammatory responses. TNF-α levels significantly increased at 5 µM but diminished at the highest concentration (10 µM) [62]. This paradoxical concentration-dependent effect suggests complex signaling dynamics in the cutaneous response to CDK4/6 inhibition.
Figure 2: Abemaciclib-Induced Cutaneous Toxicity Pathway. This diagram outlines the mechanistic pathway through which abemaciclib triggers inflammatory responses and apoptosis in keratinocytes, leading to clinical dermatological adverse reactions.
Comparative Analysis of Synthesis and Mechanism
The development histories of losartan and abemaciclib reflect evolving paradigms in pharmaceutical process chemistry. Losartan's journey showcases the industry's shift toward green chemistry principles, replacing traditional stoichiometric metallurgy with catalytic cross-coupling and eliminating toxic reagents like organotin compounds [61] [63]. The successful implementation of bio-derived PdNPs exemplifies modern sustainable catalysis applied to mature pharmaceutical compounds.
For abemaciclib, the research focus has centered on understanding its mechanistic toxicology rather than synthetic optimization. The elucidation of oxidative inflammatory pathways in keratinocytes provides a molecular foundation for managing dermatological adverse events, enabling better risk stratification and prophylactic strategies [62] [66]. This represents the growing field of pharmacological toxicology, where adverse effect mechanisms are systematically investigated to improve therapeutic indices.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Experimental Studies
| Reagent / Material | Function in Research | Application Example |
|---|---|---|
| Palladium Nanoparticles (PdNPs) | Catalyzes carbon-carbon bond formation | Suzuki–Miyaura cross-coupling in losartan synthesis [61] |
| Sargassum incisifolium Extract | Natural source of reducing/stabilizing agents for nanoparticle synthesis | Green synthesis of PdNPs [61] |
| N-Bromosuccinimide (NBS) | Electrophilic bromination source | Benzylic bromination in losartan intermediate synthesis [61] |
| Human Keratinocyte (HaCaT) Cell Line | In vitro model of human skin biology | Mechanistic studies of abemaciclib cutaneous toxicity [62] |
| Sodium Azide (NaN₃) | Tetrazole ring formation via [2+3] cycloaddition | Traditional losartan synthesis (requires careful handling) [63] |
This case study demonstrates the critical importance of reaction mechanism elucidation throughout the drug development lifecycle. For losartan, mechanistic understanding enabled the evolution from traditional stoichiometric methods to modern catalytic processes aligned with green chemistry principles. For abemaciclib, mechanistic toxicology studies revealed the inflammatory pathways underlying cutaneous adverse reactions, informing clinical management strategies. Together, these examples underscore how deep mechanistic investigation—from synthetic pathway optimization to biological response characterization—continues to drive innovations in pharmaceutical research and development, ultimately enhancing both manufacturing sustainability and patient care.
The field of organic chemistry continuously fuels drug discovery by providing novel synthetic methodologies and a deep understanding of reaction mechanisms. This knowledge forms the foundational bedrock upon which new therapeutic modalities are built. Among the most promising of these emerging modalities are PROteolysis TArgeting Chimeras (PROTACs), cyclopeptides, and DNA-Encoded Libraries (DELs). Each represents a paradigm shift, moving beyond the traditional occupancy-based inhibition to explore new pharmacological spaces. PROTACs harness the cell's own degradation machinery to remove disease-causing proteins. Cyclopeptides offer exceptional binding affinity and specificity due to their constrained structures. DELs leverage the power of molecular biology to screen libraries of unprecedented size, accelerating the discovery of hits against challenging targets. This guide delves into the core chemical mechanisms, design principles, and experimental protocols for these three modalities, framing them within the context of advanced organic and bioorthogonal chemistry for a scientific audience.
PROTACs: Targeted Protein Degradation
Core Mechanism and Design
PROteolysis TArgeting Chimeras (PROTACs) are heterobifunctional molecules that degrade target proteins by hijacking the ubiquitin-proteasome system (UPS) [67]. Unlike traditional small-molecule inhibitors that merely block protein activity, PROTACs catalytically induce the complete destruction of the protein of interest (POI) [68]. A PROTAC molecule consists of three key elements:
- A warhead that binds to the POI.
- A ligand that recruits an E3 ubiquitin ligase.
- A linker that connects these two moieties [67] [68].
The mechanism is a sophisticated application of induced proximity [67]:
- The PROTAC simultaneously engages the POI and an E3 ubiquitin ligase, forming a productive POI-PROTAC-E3 ligase ternary complex.
- The E3 ligase, now in close proximity to the POI, mediates the transfer of ubiquitin chains from an E2 conjugating enzyme onto lysine residues of the POI.
- The polyubiquitinated POI is recognized and degraded by the 26S proteasome [67] [68].
- The PROTAC is released and can catalyze multiple rounds of degradation, operating in a sub-stoichiometric manner [68].
This mechanism is visualized in the following diagram:
Key E3 Ligases and Clinical Progress
The choice of E3 ligase ligand is critical for PROTAC design. The most commonly utilized E3 ligases to date are Cereblon (CRBN) and Von Hippel-Lindau (VHL), for which small-molecule ligands have been developed [67]. The clinical translation of PROTACs is advancing rapidly, with several candidates in clinical trials.
Table 1: Representative E3 Ligases Used in PROTAC Design
| E3 Ligase | Small-Molecule Ligand | Key Characteristics |
|---|---|---|
| Cereblon (CRBN) | Thalidomide, Lenalidomide, Pomalidomide | Well-characterized; basis for IMiD drugs; recruits transcription factors like IKZF1/3 [67]. |
| Von Hippel-Lindau (VHL) | VH032 derivatives | Hydroxyproline-based ligands with nanomolar affinity; used in selective PROTACs like MZ1 [67]. |
| MDM2 | Nutlin | Used in the first all-small-molecule PROTAC targeting the Androgen Receptor (AR) [67]. |
Table 2: Selected PROTACs in Clinical Trials (as of 2022)
| PROTAC Name | Target | E3 Ligase | Clinical Phase | Indication |
|---|---|---|---|---|
| ARV-110 | Androgen Receptor (AR) | CRBN | Phase II | Prostate Cancer [67] |
| ARV-471 | Estrogen Receptor (ER) | CRBN | Phase II | Breast Cancer [67] |
Experimental Protocol: In Vitro Assessment of PROTAC Activity
Objective: To evaluate the degradation efficiency and specificity of a novel PROTAC molecule. Materials:
- Cell line expressing the target protein.
- PROTAC molecule (as a 10 mM stock in DMSO).
- Control compounds: DMSO, matching POI inhibitor (for negative control), known active PROTAC (for positive control).
- Lysis buffer (e.g., RIPA buffer with protease and phosphatase inhibitors).
- Antibodies for Western Blot: against POI, and a loading control (e.g., GAPDH, β-Actin).
- Cell culture reagents and equipment.
Method:
- Cell Seeding and Treatment: Seed cells in a 6-well or 12-well plate and allow to adhere overnight. Treat cells with a concentration series of the PROTAC (e.g., 1 nM, 10 nM, 100 nM, 1 µM) and controls for a predetermined time (typically 4-24 hours).
- Cell Lysis: Aspirate media, wash cells with PBS, and lyse cells using ice-cold lysis buffer. Incubate on ice for 10-20 minutes, then centrifuge at >14,000 g for 15 minutes at 4°C to pellet debris.
- Protein Quantification and Western Blot: Determine protein concentration of the supernatant using a BCA or Bradford assay. Prepare samples with Laemmli buffer, denature, and separate equal protein amounts by SDS-PAGE. Transfer to a PVDF membrane.
- Immunoblotting: Block the membrane, then probe with primary antibody against the POI overnight at 4°C. After washing, incubate with an HRP-conjugated secondary antibody. Develop the blot using enhanced chemiluminescence (ECL) reagent and visualize.
- Data Analysis: Quantify band intensity using image analysis software (e.g., ImageJ). Normalize POI signal to the loading control. Plot normalized POI levels versus PROTAC concentration to determine the half-maximal degradation concentration (DC₅₀).
Cyclopeptides: Constrained Structures for Enhanced Targeting
Biosynthetic Mechanisms and Chemical Advantages
Cyclopeptides, or cyclic peptides, are polypeptides whose N- and C-termini are connected via an amide bond, forming a circular backbone [69]. This cyclization confers significant advantages over their linear counterparts, including enhanced metabolic stability (resistance to exopeptidases), structural rigidity, and improved binding affinity and selectivity for target proteins [69]. Their ability to target protein-protein interactions (PPIs) makes them particularly attractive for drug discovery.
Cyclopeptides are synthesized through two primary biosynthetic pathways, each with distinct cyclization mechanisms:
- Ribosomal Synthesis and Post-Translational Modification: Linear peptide precursors are synthesized ribosomally and then cyclized by specific enzymes [70].
- Endopeptidase-catalyzed cyclization: Enzymes like Asparaginyl Endopeptidase (AEP) cyclize plant-derived cyclotides by a transpeptidation reaction. The AEP recognizes a conserved motif (e.g., XXNGLP), cleaves the precursor, and forms a new peptide bond between the N- and C-termini to create the cyclic backbone [70]. Similarly, subtilisin-like proteases (e.g., PatG) cyclize cyanobactins in a mechanism analogous to thioesterase domains in NRPSs [70].
- Non-Ribosomal Peptide Synthetases (NRPS): Large, modular enzyme complexes assemble peptides independently of the ribosome. Cyclization is typically catalyzed by a thioesterase (TE) domain located at the C-terminus of the synthetase. The TE domain facilitates nucleophilic attack by an internal amine, hydroxyl, or thiol group on the carbonyl carbon of the thioester, releasing the peptide as a cyclic structure [70].
Anticancer Applications and Key Examples
Natural cyclopeptides have demonstrated potent anticancer activities, with many novel compounds discovered in the past two decades. Their efficacy stems from diverse mechanisms, including histone deacetylase (HDAC) inhibition, ion channel modulation, and induction of apoptosis [69].
Table 3: Selected Anticancer Cyclopeptides from Natural Sources
| Cyclopeptide Name | Biological Source | Reported Anticancer Activity |
|---|---|---|
| Azumamide A | Sponge (Mycale izuensis) | HDAC inhibitory activity (IC₅₀ = 0.045 µM against K562 cells); cytostatic effects [69]. |
| Astin C | Plant (Aster tataricus) | Inhibits the cGAS-STING signaling pathway (Kd = 2.3 µM for STING), shows potential for autoimmune disease treatment [71]. |
| Homophymine A | Sponge (Homophymia sp.) | Potent anti-proliferative activity against various cancer cell lines (IC₅₀ = 2-100 nM) [69]. |
| Microsclerodermin A | Sponge (Microscleroderma herdmani) | Induces apoptosis in pancreatic cancer cells (e.g., IC₅₀ = 0.8 µM in BxPC-3) [69]. |
Experimental Protocol: Assessing Cyclopeptide Stability in Serum
Objective: To determine the metabolic stability of a synthetic cyclopeptide in serum, a key parameter for its development as a therapeutic. Materials:
- Cyclopeptide (lyophilized).
- Mouse or human serum (commercially available).
- Control linear peptide.
- Precipitation solvents: Acetonitrile or Trichloroacetic acid (TCA).
- HPLC system with UV/VIS or MS detector.
- Water bath or incubator set to 37°C.
Method:
- Sample Preparation: Pre-warm serum to 37°C. Dissolve the cyclopeptide in a minimal volume of DMSO or water and spike it into the serum to achieve a final concentration of 10-100 µM. Mix thoroughly.
- Incubation: Immediately remove a time-zero aliquot (t=0). Continue incubating the serum-peptide mixture at 37°C. Remove aliquots at predetermined time points (e.g., 0, 15, 30, 60, 120, 240 minutes).
- Protein Precipitation: For each aliquot, add 2-3 volumes of ice-cold acetonitrile to precipitate serum proteins. Vortex vigorously and incubate on ice for 10 minutes. Centrifuge at high speed (>14,000 g) for 10 minutes to pellet the protein.
- Analysis: Transfer the clear supernatant to a new vial and analyze by HPLC. Use an isocratic or gradient method suitable for resolving the peptide.
- Data Analysis: Measure the peak area of the intact cyclopeptide at each time point. Plot the natural logarithm of the peak area (or percentage remaining) versus time. The slope of the linear regression is the degradation rate constant (k). The half-life (t₁/₂) can be calculated as t₁/₂ = ln(2)/k.
DNA-Encoded Libraries (DELs): High-Throughput Discovery
DNA-Encoded Library (DEL) technology is a powerful platform that allows for the affinity-based screening of incredibly large chemical libraries (billions to trillions of compounds) in a single experiment [72] [73]. The core concept involves covalently linking each unique small molecule in a library to a unique DNA tag that serves as an amplifiable barcode for identification [72] [74].
The most common method for constructing DELs is the DNA-recorded split-and-pool synthesis [72] [73]:
- Split: A set of starting building blocks (BBs), each attached to a unique DNA tag, is divided into separate reaction vessels.
- React: A second set of BBs is coupled to the first in each vessel. The identity of this second BB is recorded by enzymatically ligating a new, unique DNA tag to the existing tag.
- Pool: All reactions are pooled together, creating a library where each compound is tagged with a DNA sequence that records its synthetic history.
- This iterative process enables the combinatorial creation of vast libraries from a relatively small set of building blocks. The workflow for DEL construction and screening is outlined below:
Key Strategies and Emerging Applications
DEL technology has evolved to include various strategies. Single-pharmacophore libraries are built via the split-and-pool method described above, where the final molecule is a single entity synthesized in multiple steps [72]. In contrast, dual-pharmacophore libraries (e.g., Encoded Self-Assembled Chemical - ESAC) involve the self-assembly of two separate DNA-conjugated fragments, which can synergistically bind to a target [73].
A significant advancement is the development of covalent DELs (CoDEL), which incorporate electrophilic warheads (e.g., Michael acceptors) to discover irreversible covalent inhibitors. Screening these libraries often involves denaturing wash steps (e.g., with SDS) to remove non-covalent binders, ensuring the identification of compounds that form covalent bonds with the target protein [75].
Experimental Protocol: Basic Affinity Selection with a DEL
Objective: To identify binders to a purified protein target from a DNA-encoded chemical library. Materials:
- DEL (typically provided as an aqueous solution).
- Purified target protein, biotinylated.
- Streptavidin-coated magnetic beads.
- Selection buffer (e.g., PBS with 0.05% Tween-20 and BSA).
- Wash buffers (e.g., selection buffer, possibly including a denaturing buffer like 1% SDS for CoDELs).
- Elution buffer (e.g., water, or a high-pH buffer).
- Thermonixer, magnetic rack, and equipment for PCR and NGS.
Method:
- Immobilization: Incubate the biotinylated target protein with streptavidin magnetic beads in selection buffer for 30-60 minutes at 4°C. Use beads without protein as a negative control to identify non-specific binders.
- Incubation with DEL: Wash the protein-bound beads to remove unbound protein. Resuspend the beads in selection buffer and add the DEL. Incubate with gentle mixing for 1-16 hours at room temperature or 4°C.
- Washing: Place the tube on a magnetic rack to capture the beads. Carefully remove the supernatant. Wash the beads multiple times (e.g., 5-10x) with selection buffer to remove unbound and weakly bound library members. For CoDEL screens, include one or more washes with a denaturing buffer (e.g., 1% SDS).
- Elution: After the final wash, elute the specifically bound library members. This can be achieved by denaturation (e.g., heating to 95°C in water or a low-salt buffer) or by proteolytic digestion of the protein target.
- Identification: Use the eluate as a template for PCR amplification of the DNA barcodes. Submit the PCR product for next-generation sequencing (NGS). The resulting sequencing data is analyzed bioinformatically; barcodes that are significantly enriched in the protein sample compared to the negative control correspond to hit compounds.
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for Featured Modalities
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| E3 Ligase Ligands | Constructing PROTAC molecules; recruiting specific E3 ligases. | VH032: A high-affinity ligand for VHL E3 ligase [67]. Pomalidomide: A CRBN E3 ligase recruiter used in PROTACs like ARV-825 [67]. |
| DNA-Compatible Building Blocks | Synthesis of DNA-Encoded Libraries (DELs). | A diverse collection of chemical fragments (e.g., carboxylic acids, amines, aldehydes) that undergo reactions compatible with the presence of DNA [72] [74]. Amgen reported a collection of ~60,000 such building blocks [74]. |
| DNA Ligases & Polymerases | Enzymatic ligation of DNA barcodes during DEL synthesis; PCR amplification of barcodes after selection. | T4 DNA Ligase for splint-mediated ligation; Klenow Fragment for gap-filling; thermostable DNA polymerase (e.g., Taq) for PCR [72]. |
| Streptavidin-Coated Magnetic Beads | Immobilization of biotinylated protein targets for DEL affinity selections. | Used to capture and wash the target protein-library complex efficiently [72] [75]. |
| Next-Generation Sequencing (NGS) | Identification of enriched DNA barcodes from DEL selections. | Essential for decoding the results of a DEL screen; enables the parallel sequencing of millions of DNA barcodes [72] [75]. |
| Endopeptidase Enzymes | Studying or catalyzing the cyclization of ribosomally synthesized cyclopeptides. | Asparaginyl Endopeptidase (AEP): Catalyzes the backbone cyclization of cyclotides [70]. PatG: A subtilisin-like protease that cyclizes cyanobactins [70]. |
PROTACs, cyclopeptides, and DNA-encoded libraries represent the vanguard of modern drug discovery, each leveraging deep mechanistic insights from organic and biological chemistry. PROTACs demonstrate the power of catalytic, event-driven pharmacology over traditional occupancy-based inhibition. Cyclopeptides exploit conformational restraint to achieve unmatched specificity and stability for engaging challenging targets like protein-protein interfaces. DELs utilize the information-encoding capacity of DNA to overcome the physical and logistical barriers of screening vast chemical space. As our understanding of the underlying reaction mechanisms—from ubiquitin transfer to enzymatic cyclization and DNA-compatible synthesis—continues to deepen, so too will our ability to refine these modalities and develop the next generation of transformative therapeutics.
The Role of Mechanism in Structure-Activity Relationship (SAR) Exploration
Structure-Activity Relationship (SAR) analysis represents a cornerstone of modern drug discovery and organic chemistry research, establishing the critical link between a molecule's chemical structure and its biological activity. The fundamental concept, first presented by Alexander Crum Brown and Thomas Richard Fraser in 1868, posits that specific modifications to a compound's structure directly influence its physiological action [76]. While traditional SAR approaches have primarily focused on empirical correlations between structural features and biological endpoints, integrating reaction mechanism understanding provides a transformative dimension to SAR exploration that moves beyond correlative relationships toward predictive, causal models.
This technical guide examines the essential integration of mechanistic organic chemistry principles into SAR analysis, framing this synthesis within the broader context of reaction mechanism research. For medicinal chemists and researchers, appreciating the underlying chemical mechanisms—including metabolic pathways, reactive intermediate formation, and structure-based reactivity predictions—enables more rational drug design and optimization strategies [77]. By elucidating how specific chemical functionalities dictate not only binding interactions but also chemical stability and metabolic fate, mechanism-informed SAR provides a powerful framework for navigating chemical space efficiently [78].
Fundamental Concepts: Bridging Reaction Mechanisms and SAR
Core Principles of SAR
The foundation of SAR analysis rests on systematic modification of molecular structures to determine the chemical groups responsible for evoking target biological effects [76]. These modifications encompass diverse structural elements, each influencing biological activity through distinct mechanistic pathways:
- Functional group modifications: Altering key functional groups can significantly impact solubility, stability, and reactivity through electronic and steric effects that modify target binding interactions [79].
- Stereochemistry and chirality: The spatial arrangement of atoms critically influences biological activity through three-dimensional complementarity with target binding sites, where different enantiomers may exhibit dramatically different therapeutic effects [79].
- Ring size and fusion effects: Structural rigidity and conformational flexibility imposed by cyclic systems affect the molecule's ability to adopt bioactive conformations, with fused rings creating unique spatial arrangements that enhance binding affinity [79].
- Electronic effects: Inductive and resonance effects transmitted through molecular frameworks influence electron distribution at key positions, modulating reactivity and binding interactions with biological targets [79].
The Mechanistic Connection
Reaction mechanism understanding provides the theoretical framework connecting molecular structure to chemical behavior in biological systems. This mechanistic perspective operates at multiple levels:
- Molecular orbital interactions that dictate reactivity patterns and susceptibility to metabolic transformation
- Transition state stabilization principles that inform the design of enzyme inhibitors
- Reactive intermediate formation that may lead to undesired covalent adducts or mechanism-based inactivation
- Electronic and steric parameters that govern reaction pathways and rates in physiological environments
The integration of these mechanistic principles transforms SAR from a purely observational tool to a predictive framework, enabling researchers to anticipate how structural modifications will influence not only binding affinity but also metabolic stability, mechanism-based toxicity, and overall drug-like behavior [78] [77].
Computational Methodologies for Mechanism-Informed SAR
Quantitative Structure-Activity Relationship (QSAR) Modeling
Quantitative Structure-Activity Relationships (QSAR) represent the mathematical formalization of SAR principles, correlating chemical structure with biological activity through statistical models [79]. These approaches range from traditional linear regression methods to advanced machine learning algorithms, each with distinct capabilities for capturing mechanistic relationships:
- Multiple Linear Regression (MLR): Correlates molecular descriptors with biological activity using linear relationships, providing interpretable models but limited capacity for complex mechanistic relationships [80].
- Artificial Neural Networks (ANN): Models complex, non-linear relationships between molecular structure and biological activity through layered computational networks, capable of capturing intricate mechanistic patterns [80].
- Support Vector Machine (SVM): Effective for classification and regression tasks in SAR analysis, particularly valuable for distinguishing active and inactive compounds based on mechanistic descriptors [80].
Table 1: Comparison of QSAR Modeling Approaches for Mechanism-Based SAR
| Model Type | Mechanistic Interpretability | Non-Linear Capability | Best Applications in Mechanism-Informed SAR |
|---|---|---|---|
| Multiple Linear Regression (MLR) | High | Limited | Linear free-energy relationships, Hammett analyses |
| Principal Component Analysis (PCA) | Moderate | Limited | Dimensionality reduction of mechanistic descriptor spaces |
| Artificial Neural Networks (ANN) | Low | High | Complex multi-parameter optimization with unknown mechanisms |
| Support Vector Machine (SVM) | Moderate | High | Classification of compounds by mechanism of action |
Advanced Mechanistic Modeling Approaches
Beyond traditional QSAR, several specialized computational approaches directly incorporate reaction mechanism principles into SAR exploration:
Structure-Activity Landscape Modeling Activity landscape approaches provide a powerful alternative view of SAR data by simultaneously considering chemical structure and biological activity in a topographic representation [78]. These landscapes reveal characteristic regions:
- Smooth regions where similar structures display similar activities, suggesting conserved binding modes and mechanism of action
- Activity cliffs where small structural changes produce large activity differences, indicating critical mechanistic transitions or binding mode changes [78]
Inverse QSAR Approaches Inverse QSAR methodologies address the challenge of identifying structures that match a desired activity profile, essentially working backward from mechanism to structure [78]. These approaches:
- Derive optimal descriptor values for target activities
- Map descriptor spaces back to chemical structures
- Enable de novo design of compounds with specified mechanistic profiles
Domain of Applicability Assessment A critical aspect of reliable SAR modeling involves defining the "domain of applicability" (DA)—the chemical space where model predictions remain reliable [78]. DA determination methods include:
- Similarity to training set molecules using appropriate distance metrics
- Ranges of descriptor values in principal component space
- Leverage and influence statistics for regression-based models
Table 2: Domain of Applicability Assessment Methods for Mechanistic SAR Models
| Method | Key Principle | Advantages | Limitations |
|---|---|---|---|
| Similarity to Nearest Neighbor | Distance to closest training set compound | Intuitive, easy to implement | Sensitive to training set diversity |
| Dimension Related Distance | Similarity to entire training set | Comprehensive assessment | Computationally intensive for large sets |
| Principal Component Ranges | Position in reduced descriptor space | Accounts for multivariate correlations | May miss local density variations |
| Leverage in Regression | Position relative to model influence points | Statistically rigorous for linear models | Limited to regression-based approaches |
Experimental Protocols for Mechanistic SAR Exploration
Protocol 1: SAR Expansion Through Scaffold Pruning and hopping
Objective: Systematically identify minimal pharmacophore requirements and explore alternative structural frameworks with conserved mechanism of action.
Materials and Reagents:
- Commercial compound databases (CAS SciFinder, ChEMBL)
- High-purity commercial analogs for experimental validation
- Analytical tools for compound verification (LC-MS, NMR)
- Target-specific bioactivity assays
Methodology:
- Iterative scaffold simplification: Systematically remove functional group substitutions from lead compound to identify core structural requirements for activity [77].
- Comprehensive analog testing: Source and test commercially available compounds containing the core scaffold with varying substitutions.
- Similarity-based scaffold hopping: Use chemical similarity searches to identify structurally similar compounds with alternative core frameworks [77].
- Mechanistic interpretation: Analyze resulting SAR to distinguish continuous "clean" SAR (suggesting conserved mechanism) from erratic SAR (suggesting mechanism hopping) [77].
Data Interpretation:
- Continuous, interpretable activity changes suggest well-behaved, on-target activity through a conserved mechanism
- Abrupt "activity cliffs" may indicate changes in binding mode or mechanism of action
- Successful scaffold hops with maintained activity suggest separation of core binding elements from structural constraints
Protocol 2: Mechanism-Based Lead Optimization
Objective: Systematically optimize lead compounds using mechanistic insights to enhance potency, selectivity, and metabolic stability.
Materials and Reagents:
- Site-directed mutagenesis kits for target protein engineering
- Crystallization trays for protein-ligand complex structure determination
- Metabolic stability assay systems (microsomes, hepatocytes)
- Molecular modeling software with docking capabilities
Methodology:
- Binding mode elucidation: Determine high-resolution structures of ligand-target complexes using X-ray crystallography or cryo-EM [80].
- Key interaction mapping: Identify critical hydrogen bonds, hydrophobic contacts, and ionic interactions contributing to binding affinity [79].
- Mechanism-based design: Introduce strategic modifications to enhance favorable interactions while addressing metabolic soft spots [80].
- Experimental validation: Test optimized compounds in functional assays to verify maintained or improved mechanism of action.
Data Interpretation:
- Enhanced potency with maintained selectivity profile suggests successful optimization of target interactions
- Improved metabolic stability without compromised activity indicates effective mechanism-based design
- Crystallographic confirmation of predicted binding modes validates mechanistic hypotheses
Mechanistic SAR Optimization Workflow
Applications in Drug Discovery and Safety Assessment
Antitarget Profiling and Toxicity Prediction
Mechanism-informed SAR plays a critical role in predicting and avoiding adverse drug reactions (ADRs) through antitarget profiling—assessing unintended interactions with biological targets associated with toxicity [81]. Key developments in this area include:
Comparative SAR/QSAR Modeling Recent comprehensive studies comparing qualitative SAR and quantitative QSAR models for antitarget prediction revealed:
- Higher balanced accuracy for qualitative SAR models (0.80-0.81) versus quantitative QSAR models (0.73-0.76) for predicting Ki and IC50 values [81]
- Superior sensitivity for SAR models but higher specificity for QSAR models in classifying compounds as active/inactive against antitargets
- Expanded applicability domains for SAR models, covering more chemical space with reliable predictions [81]
Mechanistic Toxicity Prediction Understanding the chemical mechanisms underlying toxicity enables more predictive SAR development:
- Structural features associated with metabolic activation to reactive intermediates
- Electronic parameters predicting HERG channel binding and cardiac arrhythmia risk
- Steric and hydrophobic descriptors correlating with phospholipidosis induction
Table 3: Mechanism-Based SAR Applications in Safety Assessment
| Antitarget | Associated ADRs | Key Structural Alerts | Mechanistic Basis |
|---|---|---|---|
| hERG potassium channel | Life-threatening arrhythmias | Basic amines, hydrophobic aromatics | Pore block through π-cation interactions |
| Cytochrome P450 inhibitors | Drug-drug interactions | Nitrogen heterocycles, unsubstituted imidazoles | Heme coordination, competitive inhibition |
| Phospholipidosis-inducers | Lipid accumulation | Cationic amphiphilic structures | Lysosomal phospholipase inhibition |
| Acyl glucuronide formers | Idiosyncratic toxicity | Carboxylic acids, specific ring systems | Reactive acyl glucuronide formation |
Environmental Fate and Biodegradability Assessment
Beyond pharmaceutical applications, mechanism-informed SAR principles enable prediction of chemical persistence and biodegradability in environmental systems [76]. Structure-biodegradability relationships (SBR) incorporate mechanistic understanding of:
- Electron-withdrawing substituents (e.g., halogens) that confer persistence under aerobic conditions by stabilizing against oxidative mechanisms [76]
- Molecular accessibility to enzymatic transformation based on steric and electronic parameters
- Bioavailability considerations incorporating sorption and partitioning behavior that modulates biological accessibility
Table 4: Key Research Reagent Solutions for Mechanistic SAR Exploration
| Tool/Resource | Function in SAR Exploration | Key Features |
|---|---|---|
| GUSAR Software | QSAR model development using MNA and QNA descriptors | Self-consistent regression, applicability domain assessment [81] |
| VEGA Platform | Integrated QSAR models for toxicity and environmental fate prediction | Multiple validated models, regulatory acceptance [82] |
| EPI Suite | Environmental parameter prediction | Extensive chemical fate database, well-established models [82] |
| Chemical Databases (ChEMBL, PubChem) | Source of structural and bioactivity data | Large-scale curated data, patent extracts [81] [77] |
| Reaction Mechanism Generator (RMG) | Automatic generation of reaction mechanisms | Elementary reaction step construction, kinetic modeling [83] |
| Molecular Operating Environment (MOE) | Comprehensive molecular modeling and SAR analysis | Integrated descriptor calculation, visualization [80] |
The integration of reaction mechanism principles into Structure-Activity Relationship analysis represents a paradigm shift in molecular design and optimization. This mechanistic perspective enables researchers to move beyond empirical correlation to establish causal relationships between chemical structure and biological activity, creating a more predictive and rational framework for chemical exploration. As computational methods advance and mechanistic understanding deepens, the synergy between reaction mechanism elucidation and SAR analysis will continue to accelerate the discovery and optimization of bioactive compounds with enhanced efficacy and safety profiles.
For medicinal chemists and researchers, embracing this integrated approach provides powerful strategies for navigating complex chemical spaces, anticipating metabolic vulnerabilities, and designing compounds with optimal therapeutic properties. The continued development of mechanism-informed SAR methodologies promises to further bridge the gap between fundamental organic chemistry principles and practical drug discovery applications, ultimately enhancing the efficiency and success of pharmaceutical development.
Overcoming Challenges and Optimizing Reaction Conditions
Identifying and Mitigating Side Reactions and Byproduct Formation
In organic chemistry research, the ideal reaction pathway often competes with undesired side reactions, leading to byproduct formation that can compromise yield, purity, and efficiency. This is particularly critical in pharmaceutical development, where impurities can significantly impact drug safety and efficacy. Traditionally, optimization has relied on labor-intensive, time-consuming experimentation guided by chemical intuition [40]. However, a paradigm shift is underway, enabled by advances in lab automation and the introduction of machine learning algorithms that synchronously optimize multiple reaction variables with minimal human intervention [40]. This guide examines the core principles of side reaction identification and mitigation, integrating established laboratory techniques with cutting-edge computational approaches to provide a modern framework for controlling reaction outcomes.
Fundamentals of Reaction Pathways and Byproduct Formation
Conceptualizing Parallel Reaction Networks
Every chemical transformation exists within a network of potential pathways. The primary pathway leads to the desired product, while side pathways consume starting materials or intermediates to form byproducts. These competing reactions are often driven by:
- Functional group selectivity: Reagents interacting with different functional groups present in the substrate.
- Intermediate stability: Reactive intermediates (e.g., carbocations, radicals, anions) partitioning between multiple reaction channels.
- Thermodynamic vs. kinetic control: Conditions favoring the most stable product versus the most rapidly formed product.
- Catalyst selectivity: Enzymatic or synthetic catalysts promoting specific stereoelectronic pathways.
Understanding this network is crucial for developing effective mitigation strategies, as eliminating one side reaction may inadvertently enhance another.
Analytical Techniques for Byproduct Identification
Robust analytical characterization is fundamental for identifying and quantifying byproducts.
- Chromatographic Methods: HPLC, GC (as used in Organic Syntheses procedures for establishing 97% purity [84]), and TLC provide separation and quantitative analysis of reaction mixtures.
- Spectroscopic Techniques: NMR (
^1H,^13C, 2D methods) and IR spectroscopy offer structural elucidation of isolated impurities [84] [85] [86]. - Mass Spectrometry: HRMS and LC-MS enable precise molecular weight determination and tracking of reaction pathways, even for transient species.
Computational Approaches for Reaction Network Analysis
Modern computational tools enable a priori prediction of reaction networks, providing insights before laboratory experimentation.
Automated Reaction Network Generation
Software tools like the ACE Reaction Network module in the Amsterdam Modeling Suite can automatically generate potential reaction mechanisms [87]. The process involves:
- Specifying reactants, products, and active atoms: Defining the molecular system and the atoms likely involved in bond formation/cleavage.
- Intermediate generation: Iteratively breaking and forming bonds to propose plausible intermediates.
- Network creation: Determining connections between intermediates via elementary reactions.
- Pathway minimization: Eliminating chemically implausible or high-energy pathways [87].
This computational analysis reveals not only the desired pathway but also potential side routes, allowing chemists to design strategies that avoid these pathways.
Figure 1: A generalized reaction network generated by computational analysis, showing desired and competing pathways.
Kinetic Simulation and Modeling
Powerful industrial-grade software like Ansys Chemkin and Kintecus allows for detailed modeling of complex, chemically reacting systems [88] [89]. These tools provide:
- Mechanism reduction and optimization: Identifying non-essential reactions to simplify complex models [88] [89].
- Sensitivity analysis: Determining which reactions have the greatest impact on product distribution and which rate constants require precise measurement [89].
- Uncertainty analysis: Using Monte Carlo methods to predict "real-life" behaviors with confidence intervals [89].
- Global regression/optimization: Fitting multiple datasets with different initial conditions to refine kinetic parameters [89].
Table 1: Computational Tools for Reaction Analysis and Byproduct Prediction
| Tool | Primary Function | Key Features Relevant to Byproduct Mitigation | Application Context |
|---|---|---|---|
| ACE Reaction Network [87] | Automated mechanism generation | Predicts potential side intermediates and pathways from molecular graphs | Early-stage reaction design and hypothesis generation |
| Ansys Chemkin [88] | Chemical kinetics simulation | Reaction path analysis; surrogate fuel blend formulation; mechanism reduction | Combustion, gas-phase processes, detailed chemistry applications |
| Kintecus [89] | Chemical kinetics simulation & optimization | Global data regression; uncertainty analysis; normalized sensitivity coefficients | Combustion, biological, atmospheric, and nuclear chemistry |
Experimental Methodologies for Identification and Mitigation
Protocol for Reaction Optimization Using Design of Experiments
Traditional "one-variable-at-a-time" approaches are inefficient for optimizing complex reaction systems where variables interact.
Objective: Systematically identify optimal conditions that maximize yield of the desired product while minimizing byproduct formation. Materials:
- Reaction substrates and anhydrous solvents (e.g., benzene, DMSO, dried over molecular sieves or distilled from appropriate drying agents) [84].
- Reagents and catalysts (e.g., carbodiimide resin, orthophosphoric acid) [84].
- Inert atmosphere equipment (argon or nitrogen line) [84] [86].
- Automated liquid handling system or parallel reactor block (for high-throughput).
- Analytical instrumentation (HPLC, GC, LC-MS).
Procedure:
- Define Critical Variables: Identify key factors (e.g., temperature, stoichiometry, solvent composition, catalyst loading, reaction time).
- Design Experiment Matrix: Utilize a statistical design (e.g., Full Factorial, Plackett-Burman, Central Composite Design) to define the set of experiments.
- Execute Reactions: Perform reactions in parallel under the specified conditions, ensuring meticulous control and documentation.
- Analyze Outcomes: Quantify the yield of the desired product and key byproducts for each experiment using HPLC or GC.
- Build Predictive Model: Apply regression analysis or machine learning to model the relationship between variables and outcomes.
- Validate Model: Confirm the model's accuracy by running experiments at the predicted optimum.
This methodology efficiently maps the reaction landscape, revealing interactions between variables and identifying a robust operating space that suppresses side reactions.
Case Study: Mitigation via Polymeric Reagents
The use of polymeric reagents is a strategic approach to facilitate purification and minimize byproduct contamination.
Example: Moffat Oxidation using Polymeric Carbodiimide [84]
Background: The standard Moffat oxidation uses a soluble carbodiimide, which produces dicyclohexylurea (DCU) as a byproduct that can be difficult to separate from the desired product.
Modified Procedure:
- A mixture of cis- and trans-4-tert-butylcyclohexanols (540 mg, 0.00346 mol) is dissolved in anhydrous benzene (50 mL) and DMSO (25 mL) in a 250 mL three-necked flask under argon [84].
- Polymeric carbodiimide resin (13.19 g, containing ~0.012 mol active groups) is added, followed by a DMSO solution of anhydrous orthophosphoric acid (98 mg in 0.2 mL DMSO) [84].
- The mixture is stirred at room temperature for 3.5 days [84].
- Mitigation Step: The spent resin beads are simply removed by filtration and washed with ether. The combined filtrates are washed with water to remove DMSO and concentrated to yield crude 4-tert-butylcyclohexanone in 83-84% yield and 97% purity by GC analysis [84].
Mitigation Advantage: The polymeric reagent is physically removed by filtration, eliminating the challenging DCU byproduct from the reaction mixture and greatly simplifying the workup and purification.
Protocol: In-situ IR for Reaction Monitoring
Real-time monitoring provides direct insight into reaction progression and intermediate formation.
Objective: Track the consumption of starting materials and the appearance/disappearance of intermediates and byproducts to identify optimal reaction times and quench points.
Materials:
- Reactor equipped with attenuated total reflectance (ATR) probe coupled to an FTIR spectrometer.
- Standard reaction components.
Procedure:
- Calibrate the IR method by identifying characteristic vibrational frequencies for the starting material, desired product, and known or suspected intermediates/byproducts.
- Initiate the reaction with the ATR probe immersed in the reaction mixture.
- Collect spectra at regular, short intervals (e.g., every 30-60 seconds).
- Plot the intensity (or area) of key peaks against time to generate reaction profiles.
- Use the profiles to determine the reaction endpoint, detect the accumulation of unstable intermediates, or identify the onset of decomposition pathways.
The Scientist's Toolkit: Essential Reagents and Materials
Strategic selection of reagents and materials is a primary method for controlling reaction pathways.
Table 2: Research Reagent Solutions for Byproduct Mitigation
| Reagent/Material | Function | Example Application | Mitigation Mechanism |
|---|---|---|---|
| Polymeric Carbodiimide [84] | Coupling/Dehydration Agent | Moffat oxidation of alcohols to ketones | Facilitates removal of urea byproduct via filtration, simplifying purification. |
| Molecular Sieves [84] | Water Scavenger | Reactions sensitive to hydrolysis or where water is a byproduct (e.g., acetals) | Shifts equilibrium by removing water, suppresses hydrolytic side reactions. |
| Orthophosphoric Acid [84] | Acid Catalyst | Moffat oxidation | Specific catalytic activity for the desired pathway under mild conditions. |
| Diisopropylamine [86] | Non-nucleophilic Base | Deprotonation in silicate formation | Minimizes nucleophilic attack on the electrophilic substrate, preventing alkylation byproducts. |
| Anhydrous Solvents (Benzene, DMSO, THF) [84] [86] | Reaction Medium | Moisture-sensitive reactions | Prevents hydrolysis of sensitive reagents/intermediates (e.g., acid chlorides, organometallics). |
Data Analysis and Visualization for Optimization
Effective data interpretation is critical for diagnosing and addressing side reactions.
Quantitative Analysis of Reaction Outcomes
Statistical analysis of reaction data helps move from observational to predictive understanding.
Table 3: Key Quantitative Data Analysis Methods for Reaction Optimization
| Analysis Method | Primary Use | Application in Reaction Optimization |
|---|---|---|
| Descriptive Statistics [90] | Summarize data characteristics | Calculate mean yield and standard deviation across replicate experiments to assess reproducibility. |
| Regression Analysis [90] | Model relationships between variables | Build a model linking reaction variables (T, conc.) to output (yield, impurity level). |
| Cross-Tabulation [90] | Analyze categorical relationships | Compare the frequency of a specific byproduct across different catalyst systems. |
| Sensitivity Analysis [89] | Determine parameter influence | Identify which rate constants in a mechanism most strongly affect the yield of a problematic byproduct. |
| Uncertainty Analysis [89] | Quantify confidence in predictions | Use Monte Carlo sampling to predict the range of possible impurity levels given uncertainties in rate constants. |
Visualizing Optimization Strategies
A systematic workflow integrates computational and experimental strategies for comprehensive byproduct control.
Figure 2: An integrated workflow for identifying and mitigating side reactions, combining computational and experimental methods.
The effective management of side reactions and byproducts is a cornerstone of efficient organic synthesis, especially within the demanding context of pharmaceutical research. The classical, iterative approach to reaction optimization is being superseded by an integrated strategy that leverages predictive computational tools, high-throughput experimentation, and advanced real-time analytics. This paradigm allows researchers to move from a reactive stance—addressing byproducts after they form—to a proactive one, designing reaction systems to inherently favor the desired pathway. As machine learning and automation continue to evolve [40], the ability to rapidly navigate complex chemical space and predict reaction outcomes with high accuracy will become standard practice, fundamentally advancing the design and execution of organic synthesis.
Selecting Catalysts and Ligands for Improved Efficiency and Selectivity
The selection of optimal catalysts and ligands is a cornerstone of modern organic chemistry, directly determining the efficiency and selectivity of synthetic reactions. Within the broader study of reaction mechanisms, understanding how to choose these components provides the critical link between theoretical models and practical synthetic outcomes. This guide synthesizes traditional principles with cutting-edge, data-driven approaches to equip researchers with a structured methodology for rational catalyst and ligand design. The move away from purely empirical, trial-and-error screening towards mechanism-informed and computationally assisted selection represents a paradigm shift in chemical research and development [91] [92].
Theoretical Foundations and Key Principles
The Role of Mechanisms in Catalyst Selection
A reaction mechanism provides a step-by-step map of the elementary reactions that constitute an overall chemical transformation [93]. This map is indispensable for catalyst design, as it identifies key transition states and reactive intermediates whose stability dictates the reaction rate and pathway. For catalytic cycles, mechanisms detail processes like oxidative addition, migratory insertion, and reductive elimination, pinpointing where a catalyst must interact most effectively with substrates.
The Sabatier Principle and the "Active Ligand Space"
A foundational concept for rational design is the Sabatier principle, which posits that an optimal catalyst must bind reaction intermediates neither too strongly nor too weakly [92]. Excessive binding strength poisons the catalyst, while insufficient strength fails to activate the substrate.
Quantitatively, this principle can be applied to ligand selection through the concept of a replacement energy (ΔErep). This metric compares the binding strength of a ligand (L) to that of a key reaction species (R) relative to a common reference, such as trimethyl phosphine (PMe3) [92]:
- Ligand Replacement Energy: LP + M(PMe3)2 → LPMPMe3 + PMe3, ΔErep(L)
- Reaction Species Replacement Energy: R + M(PMe3)2 → RMPMe3 + PMe3, ΔErep(R)
The difference in these energies, |ΔErep(L) - ΔErep(R)|, serves as a simple activity descriptor. The most active catalysts are found where this difference is minimized, defining a narrow Active Ligand Space (ALS)—typically within ±10 kJ mol⁻¹—where ligand and reaction species binding are optimally balanced [92]. This relationship creates a volcano-shaped plot when activity is graphed against ΔErep(L), visually encapsulating the Sabatier principle.
Computational and Data-Driven Methodologies
Modern catalyst discovery is increasingly powered by computational tools that dramatically accelerate screening and prediction.
Machine Learning and Database Construction
The establishment of specialized databases is a critical enabler for data-driven discovery. The Metal-Phosphine Catalyst Database (MPCD), for instance, is constructed through a systematic workflow [92]:
- Selection of fundamental substituent groups (e.g., alkyl, aryl, alkoxy).
- Generation of diverse P-ligands by combining these substituents.
- Assembly of ligand-metal-reference complexes (e.g., LP–M–PMe3).
- Global Potential Energy Surface (PES) exploration using methods like the stochastic surface walking (SSW) method to find the most stable conformer.
- Energy evaluation via Density Functional Theory (DFT) calculations to compute the final ΔErep(L) values.
This database allows for the rapid construction of volcano plots and the identification of the ALS for a target reaction without exhaustive experimental work [92].
Generative Models for Catalyst Design
Beyond screening known ligands, generative artificial intelligence models can propose entirely novel catalyst structures. Frameworks like CatDRX use a reaction-conditioned variational autoencoder (VAE) that learns from broad reaction databases [91]. The model integrates embeddings of the catalyst structure and reaction conditions (reactants, reagents, products) to simultaneously generate novel catalyst candidates and predict their performance (e.g., yield) [91]. This allows for the inverse design of catalysts tailored to specific reaction environments.
The following diagram illustrates the integrated human-AI workflow for catalyst discovery, from initial database construction to final experimental validation:
Quantitative Descriptors for Ligand Analysis
Effective ligand analysis relies on quantitative descriptors that capture electronic and steric properties. The table below summarizes key metrics used in rational design.
Table 1: Key Quantitative Descriptors for Ligand Analysis
| Descriptor Name | Type | Description | Role in Catalyst Design |
|---|---|---|---|
| Ligand Replacement Energy (ΔErep(L)) [92] | Electronic | Energy change for replacing a reference ligand (e.g., PMe3) with the target ligand on a metal center. | Primary metric for positioning a ligand within the Active Ligand Space (ALS) and on volcano plots. |
| Tolman Cone Angle [92] | Steric | The angular measure of the ligand's spatial footprint around the metal. | Predicts steric accessibility of the metal center; influences regioselectivity and prevents catalyst dimerization. |
| Buried Volume (%Vbur) [92] | Steric | The percentage of the metal's coordination sphere occupied by the ligand. | A more sophisticated, computed measure of steric bulk than cone angle. |
| Natural Bond Orbital (NBO) Charge [92] | Electronic | The computed charge on specific atoms (e.g., phosphorus) derived from quantum mechanical calculations. | Indicates the electron-donating or -withdrawing character of the ligand, influencing the metal's electron density. |
Experimental Protocols and Workflows
Translating computational predictions into tangible results requires rigorous experimental validation.
General Workflow for Cross-Coupling Catalyst Screening
A representative workflow for evaluating catalyst ligands in a Suzuki-Miyaura cross-coupling reaction, adapted from high-throughput screening methodologies, involves the following steps [92]:
- Reaction Setup: Conduct parallel reactions in a glovebox under an inert nitrogen atmosphere. Each reaction vessel contains the aryl halide, boronic acid, base, and the candidate metal-ligand complex in an anhydrous solvent (e.g., tetrahydrofuran).
- Reaction Execution: Stir the reaction mixture at a defined temperature for a set duration.
- Product Analysis: Use Gas Chromatography (GC) or GC-Mass Spectrometry (GC-MS) to quantify reaction conversion and yield. Compare the performance of different ligand candidates against a baseline.
- Data Integration: Correlate experimental yields with the computed ΔErep(L) values to validate the predicted volcano plot relationship and refine the ALS model.
Case Study: Synthesis of a Silicate Catalyst
The synthesis of Diisopropylammonium Bis(catecholato)cyclohexylsilicate provides a specific example of a well-documented experimental protocol for preparing a complex catalyst-relevant compound [86].
Detailed Procedure:
- Apparatus: A 250 mL, oven-dried, two-necked round-bottomed flask equipped with a magnetic stir bar, dropping funnel, and rubber septa is assembled. The system is evacuated and refilled with nitrogen three times to ensure an anhydrous, oxygen-free environment.
- Reaction: The flask is charged with pentane, anhydrous pyridine, and anhydrous methanol. The solution is cooled to 0°C, and a solution of cyclohexyltrichlorosilane in pentane is added dropwise. A voluminous white precipitate (pyridinium hydrochloride) forms.
- Workup: The mixture is stirred at room temperature for 3 hours. Upon completion, the reaction mixture is decanted from the solids, transferred to a separatory funnel, and washed with water. The organic layer is then sequentially washed with 2M aqueous HCl, saturated aqueous NaHCO₃, deionized water, and saturated aqueous NaCl.
- Isolation: The organic layer is dried over sodium sulfate, and the solvent is removed via rotary evaporation to yield the intermediate, cyclohexyltrimethoxysilane, as a clear, colorless oil in 94% yield [86].
The following diagram outlines the key stages of this synthetic protocol:
The Scientist's Toolkit: Research Reagent Solutions
Successful execution of catalytic reactions hinges on the use of high-purity, well-specified materials. The following table details essential reagents and their functions as derived from documented procedures [86] [94].
Table 2: Essential Research Reagents for Catalyst Synthesis and Screening
| Reagent / Material | Specification / Purification | Function |
|---|---|---|
| Tetrahydrofuran (THF) | Anhydrous; refluxed and distilled from sodium metal, then from lithium aluminum hydride under nitrogen [94]. | Common anhydrous solvent for organometallic reactions and catalyst preparation. |
| Trialkylboranes (e.g., BH₃•THF) | Standardized commercial solution; concentration verified by titration [94]. | Source of borane for hydroboration and the in-situ generation of organoborane species. |
| Phosphine Ligands | Commercial samples, often stored under nitrogen; may be purified by recrystallization or distillation. | Key ligand class for tuning the activity and selectivity of transition metal catalysts (e.g., Pd, Ni). |
| Pyridine | Anhydrous; stored over KOH pellets to remove water [86]. | Acts as a base and HCl scavenger in reactions involving acid chlorides or other acidic byproducts. |
| Silane Reagents (e.g., Cyclohexyltrichlorosilane) | High purity (>98%); used without further purification [86]. | Electrophilic silicon source for synthesizing silicate-based catalysts and reagents. |
| Sodium Sulfate (Na₂SO₄) | Anhydrous granules. | Drying agent for organic solutions during workup procedures. |
Advanced Catalyst Architectures
Beyond molecular catalysts, advanced materials offer unique opportunities for improving selectivity and efficiency.
Single-Atom Catalysts (SACs)
Single-Atom Catalysts (SACs), featuring isolated metal atoms on solid supports, represent a frontier in catalysis. They offer nearly 100% atom utilization, well-defined active sites, and often exceptional selectivity [95] [96]. SACs bridge the gap between homogeneous and heterogeneous catalysis [96]. When derived from Covalent Organic Frameworks (COFs) or Metal-Organic Frameworks (MOFs), SACs benefit from high surface area, structural regularity, and chemical modularity, which enhance their performance and stability in challenging reactions like CO₂ electroreduction [96].
Integrative Catalytic Pairs (ICPs)
To overcome the limitations of uniform active sites in SACs for complex reactions, Integrative Catalytic Pairs (ICPs) have been proposed. ICPs consist of spatially adjacent, electronically coupled dual active sites that function cooperatively yet independently [95]. This architecture allows for functional differentiation within a small ensemble, enabling concerted reactions involving multiple intermediates, which has shown promise in CO₂ conversion and hydrogenation reactions [95].
The field of catalyst and ligand selection is undergoing a profound transformation, driven by the integration of mechanistic chemistry, computational power, and data science. The emergence of quantitative frameworks like the Active Ligand Space, supported by expansive databases and predictive AI models, provides researchers with unprecedented tools for rational design. By grounding computational predictions in rigorous experimental protocols and leveraging novel catalyst architectures like SACs, scientists can systematically navigate the vast chemical space to discover highly efficient and selective catalytic systems, thereby accelerating research and development across the chemical and pharmaceutical industries.
Strategies for Controlling Regioselectivity and Stereoselectivity
Within the broader context of reaction mechanisms in organic chemistry research, the control of regioselectivity and stereoselectivity represents a fundamental challenge with profound implications for synthetic efficiency, particularly in pharmaceutical development. Regioselectivity refers to the preference for a reaction to occur at one atom or functional group over another that is constitutionally similar, while stereoselectivity describes the preferential formation of one stereoisomer over another [97]. These forms of selectivity are governed by the interplay between steric demands, electronic effects, and orbital interactions within reaction mechanisms [98] [99]. For researchers and drug development professionals, mastering these selectivities is not merely academic—it directly impacts drug efficacy, safety profiles, and developmental viability, as over half of therapeutic drugs are chiral and often exhibit enantiomer-specific pharmacological activities [100]. The strategic application of predictive models, detailed mechanistic understanding, and carefully designed experimental protocols enables synthetic chemists to achieve high levels of selectivity, thereby reducing wasteful byproduct formation and streamlining synthetic routes to complex target molecules.
Theoretical Foundations of Selectivity Control
Fundamental Concepts and Definitions
In organic synthesis, selectivity manifests in several distinct forms that determine the outcome of chemical transformations. Chemoselectivity describes the preferential reaction of one functional group in the presence of others, while regioselectivity addresses the preference for one structural isomer over another, such as in Markovnikov versus anti-Markovnikov addition to alkenes [97] [99]. Stereoselectivity encompasses control over the spatial arrangement of atoms in the reaction products, which can be further categorized as diastereoselectivity (preference for one diastereomer over another) or enantioselectivity (preference for one enantiomer over another) [98]. Asymmetric induction, the key principle behind stereoselectivity, can occur through internal chirality (existing within the substrate), relayed chirality (temporarily introduced and removed), or external chirality (introduced by chiral catalysts or ligands) [98]. The physical basis for these selectivities arises from energy differences between competing transition states, often measured through density functional theory (DFT) calculations that quantify activation barriers and stabilize interactions [101] [102].
Predictive Models for Stereoselectivity
Several established models predict stereochemical outcomes based on conformational analysis of transition states. The Felkin-Anh model represents a refinement of earlier Cram and Felkin models, incorporating improvements to address carbonyl stereoselectivity more accurately [98]. This model considers torsional strain, steric effects, and polar/electronic factors, particularly the antiperiplanar effect where the best nucleophile acceptor σ* orbital aligns parallel to both the π and π* orbitals of the carbonyl, providing stabilization for the incoming nucleophile [98]. The model also incorporates the Bürgi-Dunitz angle (107°), describing the non-perpendicular approach of nucleophiles to carbonyl centers [98]. For situations displaying reversed selectivity, chelation control can override traditional predictions, as seen when Lewis acids coordinate with heteroatoms in α- or β-positions, locking conformations and directing nucleophilic attack to create "anti-Felkin" products [98]. Extended models address 1,3-asymmetric induction, with the Evans model successfully predicting stereoselectivity when β-stereocenters influence carbonyl additions [98].
Table 1: Predictive Models for Stereoselectivity in Carbonyl Additions
| Model | Key Principle | Applicability | Limitations |
|---|---|---|---|
| Cram's Rule | Approach from least hindered side when carbonyl flanked by two smallest groups | Non-catalytic reactions with adjacent chiral center | Fails with eclipsed conformations and polar substituents |
| Felkin Model | Staggered transition state with large substituent perpendicular to carbonyl | Addresses torsional strain ignored by Cram | Underestimates polar effects of electron-withdrawing groups |
| Felkin-Anh Model | Incorporates antiperiplanar effect and Bürgi-Dunitz nucleophile approach | Improved prediction for aldehydes and polar substrates | Requires consideration of chelation effects separately |
| Chelation Control | Lewis acid coordination creates rigid cyclic intermediate | Substrates with α- or β-heteroatoms (O, N, S) | Requires specific metal coordination geometry |
Predictive Models for Regioselectivity
Regioselectivity is predominantly governed by electronic and steric factors that differentiate between similar reactive sites. In alkene additions, Markovnikov selectivity favors bond formation at the more substituted carbon due to stabilization of carbocation intermediates or partial positive charge development [99]. Conversely, anti-Markovnikov selectivity directs addition to the less substituted carbon, typically through alternative mechanisms such as free radical pathways or hydroboration that avoid carbocation formation [99]. For aromatic systems and complex molecules, site-selectivity becomes crucial when distinguishing between identical functional groups at different molecular positions [103]. Computational tools now provide quantitative predictions of regioselectivity by calculating activation energies for different pathways, with frontier molecular orbital (FMO) analysis particularly valuable for understanding preferences in cycloaddition and catalytic reactions [101] [102].
Computational Prediction Tools and Strategies
The emergence of sophisticated computational methods has revolutionized selectivity prediction, enabling researchers to model transition states and quantify energy differences between competing pathways before conducting experimental work.
Quantum Mechanical Approaches
Density functional theory (DFT) calculations provide fundamental insights into reaction mechanisms and selectivity by computing electronic structures and transition state geometries [101] [102]. These methods employ conceptual DFT (CDFT) indices—including electronic chemical potential (μ), chemical hardness (η), global electrophilicity (ω), and global nucleophilicity (N)—to quantify and predict reactivity patterns [102] [104]. For example, DFT studies of cobalt-catalyzed C–H functionalization of arylphosphinamides revealed that C–H cleavage and alkyne insertion steps determine stereoselectivity, while alkyne insertion alone governs regioselectivity through analysis of noncovalent interactions and frontier molecular orbitals [101]. Similarly, DFT investigations of [3+2] cycloaddition reactions for spirooxindole formation demonstrated how global reactivity indices accurately predict the nucleophilic and electrophilic character of components, enabling rational design of reactions with complete regio- and stereocontrol [102] [104]. The activation strain model and distortion/interaction analysis further decompose energy barriers into geometric distortion and electronic interaction components, providing physical explanations for unusual selectivity patterns, such as the abnormal regioselectivity observed in hexafluoropropylene oxide ring-opening reactions [105].
Machine Learning and Predictive Models
Machine learning (ML) has dramatically advanced selectivity prediction by leveraging large experimental and computational datasets to build quantitative structure-selectivity relationships. As summarized in a 2025 review, these tools span various reaction classes and implement diverse featurization techniques and model architectures [103].
Table 2: Computational Tools for Predicting Regio- and Site-Selectivity
| Tool Name | Reaction Type | Model Type | Application |
|---|---|---|---|
| pKalculator | C–H deprotonation | SQM and LightGBM | Predicting pKa and deprotonation sites |
| RegioSQM | SEAr | Semi-empirical quantum mechanics | Electrophilic aromatic substitution |
| RegioML | SEAr | LightGBM | Electrophilic aromatic substitution |
| ml-QM-GNN | Aromatic substitution | Graph neural network | Combined ML and quantum features |
| Molecular Transformer | General reaction prediction | Transformer | Broad synthesis prediction |
| Radical C-H substitution | Radical reactions | Random forest | Selectivity in radical processes |
These computational tools have become indispensable for synthetic planning, particularly in pharmaceutical research where they help prioritize synthetic routes and predict potential regioisomeric impurities. The integration of ML models with traditional quantum mechanical calculations represents the current state-of-the-art, combining the speed of pattern recognition with the fundamental physical insights of quantum chemistry [103].
Diagram 1: Computational Selectivity Prediction Workflow. This flowchart illustrates the integrated computational-experimental approach for predicting and optimizing reaction selectivity, combining quantum mechanical calculations with machine learning models.
Experimental Control Strategies and Protocols
Catalyst-Controlled Selectivity
Transition metal catalysts provide powerful means for controlling both regioselectivity and stereoselectivity through precise manipulation of coordination geometries and electronic properties. In cobalt-catalyzed C–H functionalization of arylphosphinamides, the chiral environment created by the catalyst ligand sphere directs stereoselective outcomes, with computational studies identifying C–H cleavage and alkyne insertion as the stereoselectivity-determining steps [101]. The pronounced S-selectivity in these systems arises from a larger number of noncovalent interactions in the low-energy transition state compared to higher-energy alternatives [101]. Ligand design principles enable fine-tuning of these interactions, with steric bulk, electronic donation/withdrawal, and chiral elements strategically incorporated to maximize selectivity. For industrial applications, particularly in pharmaceutical synthesis, organocatalysts and chiral Lewis acid catalysts offer complementary approaches, often providing superior selectivity for specific transformation classes while avoiding transition metal residues in final products [102].
Substrate-Controlled Selectivity
Strategic substrate design leverages inherent structural features to direct regioselectivity and stereoselectivity. The use of chiral auxiliaries represents a well-established approach, where a temporary chiral moiety is covalently attached to the substrate to direct diastereoselective formation of new stereocenters, after which the auxiliary is removed and typically recycled [98]. For regiocontrol, protecting groups can block reactive sites, while directing groups can steer catalytic transformations to specific molecular positions, as demonstrated by the phosphinamide group in cobalt-catalyzed C–H functionalization [101]. In [3+2] cycloadditions, the inherent polarization of nitrones combined with substituted nitroalkenes ensures complete regio- and stereocontrol, with the nitro group subsequently serving as a handle for further functionalization [104]. These substrate-based strategies often complement catalyst control, with the optimal approach depending on molecular complexity, synthetic step count, and overall efficiency considerations.
Reaction Condition Optimization
Beyond catalyst and substrate design, careful manipulation of reaction conditions provides additional levers for selectivity control. Solvent effects can dramatically influence selectivity by stabilizing specific transition states through polar interactions, hydrogen bonding, or coordination, as demonstrated by improved diastereoselectivity in chloroform for spirooxindole-forming [3+2] cycloadditions [102]. Temperature modulation affects selectivity when competing pathways have different activation energies, with lower temperatures typically enhancing selectivity by magnifying relative rate differences. Additives—including salts, Lewis acids, and Bronsted acids/bases—can further refine selectivity by modifying transition state energies, disrupting aggregation, or participating in coordinated delivery of reactants. High-throughput experimentation facilitates rapid screening of these multidimensional condition spaces, identifying optimal combinations that maximize selectivity while maintaining reaction efficiency [103].
Detailed Experimental Protocols
Protocol: Full Regio- and Stereoselective [3+2] Cycloaddition
This protocol outlines the synthetic procedure for achieving complete regio- and stereocontrol in the [3+2] cycloaddition between Z-C-(3-pyridyl)-N-methylnitrone and E-2-R-nitroethenes, adapted from established methodology with integration of computational validation [104].
Table 3: Research Reagent Solutions for [3+2] Cycloaddition
| Reagent | Function | Preparation & Handling |
|---|---|---|
| Z-C-(3-pyridyl)-N-methylnitrone | Nitrone 1,3-dipole component | Synthesize according to literature; store under inert atmosphere at -20°C |
| E-2-R-nitroethenes | Dipolarophile component | Recrystallize from ethanol before use; protect from light |
| Anhydrous Benzene | Reaction solvent | Dry over molecular sieves; degas with nitrogen before use |
| Ethanol (HPLC grade) | Crystallization solvent | Use without further purification |
| Deuterated Chloroform | NMR analysis | Store with molecular sieves to prevent acid formation |
Step-by-Step Procedure:
- Reaction Setup: In a flame-dried Schlenk flask under nitrogen atmosphere, dissolve Z-C-(3-pyridyl)-N-methylnitrone (1.0 mmol) in anhydrous benzene (10 mL). Add the E-2-R-nitroethene derivative (2.0 mmol) in one portion. The 1:2 molar ratio ensures complete conversion of the nitrone.
- Reaction Monitoring: Stir the reaction mixture at room temperature and monitor by TLC (silica gel, ethyl acetate/hexane 1:1) and HPLC at 12, 18, and 24 hours. The reaction typically reaches completion within 24 hours.
- Workup Procedure: After confirming complete consumption of the nitrone (TLC), concentrate the reaction mixture under reduced pressure using a rotary evaporator.
- Product Isolation: Dissolve the crude residue in minimal hot ethanol (approximately 2-3 mL) and allow slow crystallization at room temperature, then at 4°C for 12 hours. Collect the crystals by vacuum filtration and wash with cold ethanol (1 mL).
- Structural Validation: Confirm product structure by HPLC-MS and 1H/13C NMR spectroscopy. For the model reaction with E-1-nitroprop-1-ene, characteristic NMR signals include: methyl group (2.74 ppm), azolidine ring protons forming an AMX spin system (4.19-5.54 ppm), and pyridine ring protons (7.33-8.65 ppm). The key coupling constants J(H4-H5) = 4.51 Hz (trans relationship) and J(H3-H4) = 8.07 Hz (cis relationship) confirm the 3,4-cis-4,5-trans relative stereochemistry.
- Computational Validation: Perform DFT calculations at the ωB97X-D/6-31G(d,p) level to confirm the reaction mechanism and analyze electron localization function (ELF) for electron density redistribution during the reaction.
Troubleshooting Notes: If crystallization fails, purify by flash chromatography on silica gel (ethyl acetate/hexane gradient). If regioselectivity is incomplete, pre-compute global electrophilicity indices for both components to ensure appropriate pairing (nitrone ω ≈ 1.0 eV; nitroethene ω > 1.5 eV optimal).
Protocol: Computational Analysis of Selectivity
For researchers planning new selective transformations, this protocol outlines the computational assessment of potential substrates and reaction pathways.
Computational Methods:
- Geometry Optimization: Perform initial molecular geometry optimization using Gaussian 16 at B3LYP/6-31G(d,p) level for all starting materials and potential products.
- Transition State Modeling: Locate transition states for competing pathways using the QST2 or QST3 methods, followed by frequency calculations to confirm first-order saddle points (one imaginary frequency).
- Energy Calculations: Refine single-point energies at higher theory levels (e.g., ωB97X-D/6-31+G(d,p)) with solvation models (SMD or PCM) appropriate to the reaction solvent.
- Reactivity Descriptors: Calculate global reactivity indices—electronic chemical potential (μ), chemical hardness (η), global electrophilicity (ω), and global nucleophilicity (N)—from the HOMO and LUMO energies of optimized structures.
- Activation Strain Analysis: Perform distortion/interaction analysis to decompose activation energies into strain and interaction components, identifying dominant factors controlling selectivity.
- ELF Analysis: Conduct electron localization function analysis to characterize electron density redistribution and confirm mechanistic details.
Interpretation Guidelines: Compare activation barriers (ΔG‡) for competing pathways; differences > 2 kcal/mol typically yield high selectivity (>90:10). For electrophile-nucleophile pairings, optimal electrophilicity-nucleophilicity differences should exceed 0.8 eV. Use NBO analysis to identify stabilizing orbital interactions that control stereoselectivity.
Diagram 2: Stereoselectivity Determination Through Transition States. This diagram illustrates how energy differences between competing transition states (Felkin-Anh vs. Anti-Felkin) determine the major and minor product ratio in stereoselective reactions.
Pharmaceutical Applications and Significance
The control of regioselectivity and stereoselectivity has profound implications in pharmaceutical research and development, where molecular structure directly dictates biological activity. The trend toward single-enantiomer drugs has accelerated as regulatory agencies recognize the distinct pharmacological, toxicological, and metabolic profiles of individual stereoisomers [100]. Among 127 new molecular entities approved by the FDA between 2010-2014, 64% were chiral, with single enantiomers comprising the great majority (81 out of 127) [100]. Stereoselective metabolism further underscores this importance, as cytochrome P450 enzymes and UDP-glucuronosyltransferases frequently exhibit substantial stereochemical preferences, leading to different clearance rates and potential drug-drug interactions between enantiomers [100]. For example, the proton pump inhibitor omeprazole demonstrates substrate stereoselectivity in metabolism, with CYP3A4 predominantly metabolizing the (S)-enantiomer to omeprazole sulfone, while CYP2C19 primarily metabolizes the (R)-enantiomer to hydroxyomeprazole [100]. These metabolic differences result in significantly higher oral bioavailability for (S)-omeprazole (esomeprazole), which was subsequently developed as a separate drug with improved efficacy [100]. Similar considerations apply to regioselectivity, where regioisomeric impurities may exhibit unexpected toxicities or compromise therapeutic efficacy, driving the implementation of rigorous analytical control strategies throughout drug development.
The strategic control of regioselectivity and stereoselectivity represents a cornerstone of modern organic synthesis, with particular significance in pharmaceutical research where structural precision determines biological outcomes. This technical guide has outlined the integrated theoretical, computational, and experimental approaches that enable researchers to predict and achieve high levels of selectivity in synthetic transformations. The continued advancement of computational tools—particularly machine learning models trained on expansive reaction datasets—promises to further accelerate the design of selective reactions, reducing empirical optimization and expanding accessible chemical space [103]. For drug development professionals, mastery of these selectivity control strategies enables more efficient synthesis of target molecules, rational optimization of metabolic profiles, and mitigation of toxicity risks associated with isomeric impurities. As the field progresses, the integration of predictive computational methods with high-throughput experimental validation will continue to refine our understanding of reaction mechanisms and provide increasingly sophisticated tools for controlling molecular structure with precision.
Solvent and Temperature Effects on Reaction Pathway and Rate
The precise control of chemical reactions is a cornerstone of organic chemistry research, with solvent and temperature representing two of the most powerful adjustable parameters for influencing reaction pathway and rate. Within the broader context of a thesis on reaction mechanisms, understanding these effects transitions from empirical observation to predictive science, enabling researchers to rationally design conditions for synthetic efficiency, selectivity, and sustainability. For researchers and drug development professionals, this control is paramount; it dictates yield, minimizes byproducts, and ensures the scalability of synthetic routes to active pharmaceutical ingredients (APIs). This guide synthesizes contemporary research and advanced methodologies to provide a technical framework for leveraging solvent and temperature effects in mechanistic analysis and reaction optimization.
Theoretical Foundations
The Role of Temperature in Reaction Kinetics
The influence of temperature on reaction rates is traditionally described by the Arrhenius equation, ( k = A \exp(-Ea / RT) ), which establishes an exponential relationship between the rate constant ((k)) and absolute temperature ((T)). The activation energy ((Ea)) represents the energy barrier that must be overcome for the reaction to proceed. Recent studies have pushed the boundaries of this relationship, demonstrating that reactions previously considered inaccessible due to high activation barriers (50–70 kcal mol⁻¹) can be achieved under high-temperature conditions (up to 500 °C) in solution, yielding products in as little as five minutes [106]. This high-temperature synthesis expands the scope of accessible transformations, enabling pathways that are unattainable under conventional conditions.
For liquid-phase reactions, particularly at elevated temperatures approaching the solvent's critical point, the conventional Arrhenius model often fails to capture observed non-linear behavior. A new modified Arrhenius equation has been developed to account for these complexities by incorporating solvation effects on the free energy of activation: [ k{liq} = A \exp\left(-\frac{Ea + \Delta \Delta G^{\ddagger}{solv}}{RT}\right) ] where (\Delta \Delta G^{\ddagger}{solv}) represents the differential solvation effect between the transition state and reactants [107]. This model employs a minimal set of parameters to accurately describe rate constants from ambient conditions up to the critical temperature of the solvent.
Solvent Effects on Reaction Mechanisms
Solvents influence reactions through a multitude of interactions, broadly categorized as polarity/polarizability effects, specific solvent-solute interactions (e.g., hydrogen bonding), and bulk solvent properties (e.g., surface tension). The polarity of a solvent can stabilize or destabilize charged or dipolar transition states relative to reactants, thereby altering the activation barrier. A key quantitative approach describes the solvent effect through the relationship: [ \Delta G^{\ddagger}{solv} = \Delta G^{\ddagger}{gas} + \delta \Delta G^{\ddagger}{solvation} ] where (\delta \Delta G^{\ddagger}{solvation}) is the difference in solvation free energy between the transition state and the reactants [107].
In enzymatic and biomimetic systems, solvents create microenvironments that profoundly influence reactivity. The biological relevance of solvent effects cannot be overstated, as most biochemical processes occur in aqueous environments or at lipid-water interfaces. In drug development, understanding these effects is crucial for predicting in vivo behavior of drug molecules and their metabolic pathways.
Table 1: Fundamental Solvent Parameters and Their Chemical Significance
| Parameter | Description | Role in Reaction Kinetics |
|---|---|---|
| Dielectric Constant | Measure of a solvent's polarity and ability to screen electrostatic interactions | Governs stabilization of charged transition states; influences reaction rate and mechanism in polar reactions [107]. |
| Hydrogen Bond Donor/Acceptor Ability | Quantifies a solvent's capacity for specific hydrogen-bonding interactions | Can stabilize or destabilize reactants and transition states; significantly impacts reactions involving H-bonding species. |
| Polarity/Polarizability (π*) | Measures the solvent's ability to stabilize charge through dipole-induced dipole interactions | Affects rates of reactions involving dipolar or polarizable species; key in SN2 and cycloaddition reactions. |
| Electrophilicity | Characterizes the solvent's susceptibility to nucleophilic attack | Critical for reactions involving nucleophilic solvents (e.g., hydrolysis in water). |
Quantitative Data and Trends
Temperature-Dependent Kinetics in Gas and Liquid Phases
The quantitative impact of temperature is system-dependent. For the atmospheric reaction of CF₃CHO with OH radicals, kinetic studies across 204–361 K yield the temperature-dependent rate expression: [ k_1(T) = (3.8 \pm 0.2) \times 10^{-13} \times (T/300)^2 \times \exp[(131 \pm 16)/T] ] This results in a room-temperature rate constant of (5.8 \times 10^{-13}) cm³ molecule⁻¹ s⁻¹ and a atmospheric lifetime of CF₃CHO that varies from 22 days at the surface to 30 days in the colder upper troposphere [108]. This negative temperature dependence contrasts with the accelerated rates observed in high-temperature organic synthesis.
In liquid-phase systems, the deviation from Arrhenius behavior can be significant. The following table summarizes experimental data for different reaction types, highlighting the non-linear kinetics at elevated temperatures.
Table 2: Experimental Kinetic Data Showcasing Temperature and Solvent Effects
| Reaction System | Conditions | Observed Rate Constant / Yield | Interpretation |
|---|---|---|---|
| Isomerization of N-substituted pyrazoles [106] | High-temperature (up to 500 °C), p-xylene solvent | ~50% yield in 5 minutes for 50–70 kcal mol⁻¹ barriers | Demonstrates accessibility of extremely high activation barriers via high-temperature synthesis. |
| CF₃CHO + OH [108] | 204 K, gas phase | Rate constant lower than at 300 K | Exhibits negative temperature dependence, described by a modified Arrhenius expression. |
| Hydrolysis in sub-/super-critical water [107] | Wide temperature range up to critical point (Tc) | Non-Arrhenius behavior; rate "acceleration" or "slowing-down" near Tc | attributed to dramatic changes in solvent properties (e.g., dielectric constant) affecting (\Delta \Delta G^{\ddagger}_{solv}). |
| 1-Pentene Catalytic Cracking [109] | Confined ZSM-5 catalyst, 600–700 °C | Product distribution (P/E ratio) shifts with temperature | Indicates a change in dominant mechanism from carbocationic β-scission at lower T to radical-involved pathways at high T. |
Advanced Kinetic Modeling
For complex reaction networks, such as the catalytic cracking of 1-pentene over ZSM-5 at 600–700 °C, lumped kinetic models are essential. A nine-lump model, developed using the Delplot product-ordering analysis method, can successfully estimate 18 kinetic parameters, providing a theoretical basis for reactor design [109]. These models integrate the effects of temperature, catalyst properties, and feedstock to predict product selectivity, which is crucial for industrial process optimization.
Experimental and Computational Methodologies
Protocols for Kinetic Studies
A. High-Temperature Solution-Phase Kinetics [106]
- Objective: To study reactions with high activation barriers (50–70 kcal mol⁻¹).
- Methodology: Reactions are conducted in sealed glass capillaries using p-xylene as a solvent, capable of withstanding temperatures up to 500 °C. This setup allows for rapid heating and short reaction times (minutes).
- Analysis: Product composition and yield are determined using standard analytical techniques (e.g., GC-MS, NMR). Kinetic studies are complemented by DFT calculations to validate the reaction mechanism and activation barriers.
- Key Consideration: This methodology is environmentally friendly due to small scale and avoids specialized high-pressure equipment.
B. Temperature-Dependent Gas-Phase Kinetics [108]
- Objective: To determine rate coefficients ((k(T))) over a wide temperature range (e.g., 204–361 K).
- Methodology: Pulsed Laser Photolysis–Pulsed Laser Induced Fluorescence (PLP–PLIF) under pseudo-first-order conditions.
- Pulsed Laser Photolysis: A short laser pulse generates a known concentration of OH radicals.
- Pulsed Laser Induced Fluorescence: A second, time-delayed laser probes the OH concentration by exciting its fluorescence.
- Concentration Measurement: Reactant concentration (e.g., CF₃CHO) is measured in situ via Fourier-Transform Infrared (FTIR) spectroscopy.
- Data Analysis: The decay of [OH] is monitored, and the observed pseudo-first-order rate coefficient ((k')) is plotted against [reactant]. The slope of this plot gives the bimolecular rate constant (k(T)).
C. Relative-Rate Experiments [108]
- Objective: An alternative method for determining rate coefficients at a single temperature.
- Methodology: Conducted in an atmospheric simulation chamber. The decay of the reactant of interest (e.g., CF₃CHO) is measured relative to a reference compound (e.g., C₂H₆) with a well-known rate constant for reaction with OH. In-situ FTIR monitors concentrations.
- Analysis: The rate coefficient (k{\text{analyte}}) is determined from the slope of (\ln([\text{analyte}]0/[\text{analyte}]t)) versus (\ln([\text{reference}]0/[\text{reference}]_t)).
Computational Exploration of Pathways
Advanced computational programs like ARplorer are now automating the exploration of reaction pathways on potential energy surfaces (PES) [110]. ARplorer integrates quantum mechanics (QM) with rule-based methodologies, underpinned by a Large Language Model (LLM)-assisted chemical logic. Its workflow involves:
- Active Site Identification: Analyzing input molecular structures to identify reactive sites.
- Transition State Search: Iteratively optimizing structures to locate transition states using active-learning sampling.
- Pathway Validation: Performing Intrinsic Reaction Coordinate (IRC) analysis to confirm the pathway connecting reactants, transition states, and intermediates. This approach, which can use semi-empirical (GFN2-xTB) or higher-level (DFT) methods, efficiently filters out unlikely pathways, accelerating the study of complex multi-step reactions relevant to pharmaceutical chemistry.
Diagram 1: Automated computational workflow for mapping reaction pathways, integrating LLM-guided chemical logic with QM calculations [110].
Modeling Solvent Effects with Machine Learning
Machine Learning Potentials (MLPs) are emerging as powerful tools for modeling explicit solvent effects without the prohibitive cost of ab initio molecular dynamics (AIMD) for large systems [111]. An efficient active learning (AL) workflow can train reactive MLPs using only ~600 configurations, far fewer than traditional methods. The key innovations are:
- Descriptor-Based Selectors: Using Smooth Overlap of Atomic Positions (SOAP) descriptors to evaluate how well new configurations cover the PES in a feature space, ensuring data efficiency.
- Modular Training: Combining smaller datasets that separately represent the intrinsic reaction, solute-solvent, and solvent-solvent interactions.
This approach provides atomic-level insight into how explicit solvent molecules influence reaction barriers and mechanisms, moving beyond continuum models.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents and Materials for Studying Solvent and Temperature Effects
| Reagent / Material | Function and Application |
|---|---|
| p-Xylene [106] | High-boiling solvent for high-temperature (up to 500 °C) solution-phase reactions, enabling study of high-barrier transformations. |
| Sealed Glass Capillaries [106] | Reaction vessels for high-temperature synthesis; contain reactions safely at elevated temperatures and pressures. |
| Deuterated Solvents (e.g., CDCl₃, D₂O) | NMR spectroscopy for reaction monitoring and product characterization in various solvent environments. |
| ZSM-5 Zeolite Catalyst [109] | Solid acid catalyst for studying temperature-dependent mechanistic shifts (e.g., carbocationic vs. radical pathways) in hydrocarbon cracking. |
| Reference Compounds (e.g., C₂H₆) [108] | Used in relative-rate kinetic experiments to determine rate coefficients for reactions with OH radicals or other reactive species. |
Implications for Pharmaceutical Research
The principles and methodologies outlined herein have direct and profound implications for drug development. Green synthesis paradigms, including solvent-free and catalyst-free reactions, are gaining traction for their role in reducing waste and simplifying purification, aligning with the pharmaceutical industry's drive toward sustainable practices [112].
The ability to predict and control reaction pathways through solvent and temperature manipulation is critical for optimizing the synthesis of complex drug molecules. Understanding these effects aids in:
- Minimizing Degradation: Predicting and avoiding thermal degradation pathways.
- Controlling Stereoselectivity: Using solvent effects to influence the stereochemical outcome of key bond-forming steps.
- Polymorph Screening: Utilizing solvent and temperature to selectively crystallize the desired active pharmaceutical ingredient (API) polymorph.
Furthermore, the study of atmospheric reactions of fluorinated compounds, such as the formation of trifluoroacetic acid (TFA) from CF₃CHO, is directly relevant to the environmental fate and impact of fluorinated pharmaceuticals and agrichemicals [108].
Diagram 2: Integrated workflow for pharmaceutical reaction optimization, combining computational and experimental approaches to control solvent and temperature effects.
The pursuit of new therapeutic entities demands a delicate balance between biological potency and molecular properties that dictate favorable pharmacokinetics. Among these properties, lipophilicity and the Fraction of sp3 carbon atoms (Fsp3) have emerged as critical parameters influencing drug developability. Excessive lipophilicity often leads to poor aqueous solubility, limited oral bioavailability, increased metabolic clearance, and heightened risk of promiscuity and toxicity [113]. Concurrently, Fsp3, defined as the number of sp3-hybridized carbons divided by the total carbon count, has gained recognition as a vital indicator of molecular complexity and saturation [114]. Higher Fsp3 values correlate with improved solubility, enhanced success in clinical development, and reduced compound attrition rates. Within the context of organic reaction mechanisms, synthetic chemists are now tasked with developing strategic bond-forming reactions that not only build molecular complexity but also consciously modulate these crucial physicochemical parameters. This whitepaper provides a comprehensive technical guide for addressing these developability challenges through innovative synthetic methodology, analytical characterization, and computational prediction.
Theoretical Foundation: Lipophilicity and Fsp3 as Key Molecular Descriptors
The Lipophilicity-PPBS Relationship Spectrum
Lipophilicity represents a molecule's affinity for a lipophilic environment versus an aqueous one, fundamentally influencing its absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile [113]. This parameter dictates passive cellular membrane penetration, with balanced lipophilicity being essential for optimal drug action. Compounds with insufficient lipophilicity may demonstrate inadequate membrane permeability, particularly problematic for central nervous system (CNS) targets where blood-brain barrier penetration is required. Conversely, highly lipophilic molecules often exhibit poor aqueous solubility, increased plasma protein binding (PPB), and heightened risk of off-target interactions [113].
The relationship between lipophilicity and plasma protein binding represents a particularly critical consideration. As lipophilicity increases, binding to human serum albumin (HSA) and α-1-acid glycoprotein (AGP) typically intensifies, reducing the free fraction of drug available for therapeutic action. Excessive PPB (typically >95%) can substantially diminish efficacy despite high total plasma concentrations [113]. Therefore, understanding and modulating this lipophilicity-PPBS (plasma protein binding spectrum) relationship is fundamental to successful drug development.
Fsp3: Beyond Flatland Chemistry
The fraction of sp3-hybridized carbons (Fsp3) has emerged as a crucial parameter for drug-likeness, calculated as the ratio of sp3-hybridized carbons to total carbon count [114]. This metric transcends simple hydrophobicity measurements by capturing molecular complexity and three-dimensionality. Higher Fsp3 values typically correlate with:
- Improved aqueous solubility due to reduced crystal lattice energy
- Enhanced clinical success rates as demonstrated by retrospective analyses
- Reduced metabolic vulnerability due to decreased aromatic ring content
- Greater stereochemical complexity enabling more specific target engagement
Notably, spirocyclic scaffolds represent particularly valuable structural motifs due to their inherent rigidity and potential for high Fsp3, potentially offering a new generation of drug candidates with superior physicochemical profiles [114].
Analytical Methodologies for Lipophilicity and PPB Assessment
Chromatographic Determination of Lipophilicity
The International Union of Pure and Applied Chemistry (IUPAC) recommends reversed-phase chromatographic techniques for reliable lipophilicity determination [113]. These methods offer significant advantages over traditional shake-flask approaches, including minimal compound consumption, impurity tolerance, and high throughput capability.
Table 1: Chromatographic Methods for Lipophilicity Determination
| Method | Principle | Advantages | Key Applications |
|---|---|---|---|
| Reversed-Phase TLC (RP-TLC) | Partition between non-polar stationary phase and polar mobile phase | Simplicity, cost-efficiency, reduced solvent consumption | Initial lipophilicity screening, compound ranking |
| Reversed-Phase HPLC (RP-HPLC) | High-resolution separation with various detection methods | Precision, automation capability, method robustness | High-accuracy measurements, regulatory submissions |
| High Performance Affinity Chromatography (HPAC) | Retention on stationary phases immobilized with plasma proteins | Direct PPB assessment, physiological relevance | HSA/AGP binding affinity, drug-drug interaction potential |
For RP-TLC analysis, the following experimental protocol is recommended:
- Stationary Phase: RP-18W F254s aluminum plates
- Sample Preparation: Dissolve compounds in MeOH at ~0.5 mg/mL concentration
- Application: Spot 1.0 μL volumes using micropipettes
- Mobile Phase: Organic modifier/water mixtures with acidification (e.g., formic acid)
- Development: Utilize vertical developing chambers under saturated conditions
- Detection: UV visualization at 254 nm
- Calculation: Determine RМ values using RM = log(1/RF - 1) [113]
The choice of organic modifier significantly influences retention behavior. Methanol, acetonitrile, dioxane, and acetone provide varying proton-donating capabilities and polarities, enabling comprehensive lipophilicity profiling.
Plasma Protein Binding Assessment
Determining drug-plasma protein interactions represents a critical step in early discovery. High Performance Affinity Chromatography (HPAC) utilizing stationary phases with immobilized HSA or AGP has emerged as a robust, automated approach [113]. The experimental workflow includes:
- Column Selection: HSA- or AGP-immobilized silica columns
- Mobile Phase: Phosphate buffer (pH 7.4) to simulate physiological conditions
- Detection: UV detection at compound-specific wavelengths
- Interpretation: Strongly binding compounds exhibit longer retention times
Complementary methods include equilibrium dialysis, ultrafiltration, and ultracentrifugation, though these often suffer from lower throughput and reproducibility issues compared to chromatographic approaches [113].
Synthetic Strategies for Lipophilicity Reduction and Fsp3 Enhancement
Modern C(sp3)–H Functionalization Approaches
Molecular electrocatalysis provides a sustainable platform for C(sp3)–H activation through catalyst-controlled electron or atom transfer under mild conditions [115]. Both transition metal-based and metal-free catalytic systems have demonstrated efficiency in selective C(sp3)–H functionalization, offering mechanistic pathways including outer-sphere and inner-sphere electron transfer. These approaches enable unprecedented C–C bond formation while maintaining or increasing Fsp3 character through functionalization rather than simplification of complex molecular architectures.
Recent advances in photoelectrocatalysis integrate electrochemical redox control with photochemical excitation, creating synergistic activation modes for challenging C–H transformations [115]. These methods provide exceptional control over regioselectivity and stereoselectivity in asymmetric radical transformations, enabling direct installation of sp3-rich fragments without prefunctionalization.
Advanced All-Carbon Quaternary Center Construction
The formation of all-carbon quaternary centers represents a particular challenge in synthetic organic chemistry, yet provides a powerful strategy for increasing Fsp3 and structural complexity [116]. Modern approaches have evolved significantly from classical stoichiometric methods to catalytic strategies with improved efficiency and selectivity.
Table 2: Modern Methods for All-Carbon Quaternary Center Formation
| Methodology | Key Features | Fsp3 Impact | Lipophilicity Consideration |
|---|---|---|---|
| Photocatalytic SH2 radical sorting | Mild conditions, radical intermediate | High (aliphatic centers) | Tunable via radical precursor |
| Decarboxylative cross-coupling | Redox-active esters, radical capture | Moderate to High | Carboxylic acid precursors reduce lipophilicity |
| Metal-catalyzed desymmetrization | Asymmetric induction, atom economy | High (increased complexity) | Strategic introduction of polarity |
| Electrochemical processes | Oxidant-free, sustainable | Variable | Electron-rich groups often less lipophilic |
Notably, Fe(II) porphyrin-catalyzed decarboxylative cross-coupling of redox-active esters with alkenes demonstrates exceptional functional group tolerance while constructing sterically congested quaternary centers [116]. This approach exemplifies the modern paradigm of combining radical-based bond formation with transition metal catalysis to access previously challenging chemical space with high Fsp3 character.
The following diagram illustrates the strategic workflow for addressing developability issues through synthetic design:
Spirocyclic Scaffolds as Strategic Fsp3-Enhancing Motifs
Spirocyclic systems represent particularly valuable structural motifs for addressing developability challenges through their inherent rigidity and high Fsp3 character [114]. These scaffolds offer:
- Reduced molecular flexibility leading to improved target selectivity
- Enhanced three-dimensional character without excessive molecular weight
- Favorable physicochemical properties including improved solubility
- Metabolic stability compared to flat aromatic systems
Recent methodological advances have enabled more efficient access to spirocyclic architectures through catalytic asymmetric approaches, including desymmetric cycloadditions and cycloisomerization reactions [116].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential Research Reagents for Developability Optimization
| Reagent/Category | Function | Application Context |
|---|---|---|
| Redox-Active Esters (RAEs) | Radical precursors via decarboxylation | Quaternary center formation, fragment coupling |
| Fe(II) Porphyrin Complexes | Dual radical generation/capture catalyst | Decarboxylative cross-coupling reactions |
| HSA-Immobilized Columns | Plasma protein binding assessment | HPAC analysis of PPB potential |
| RP-18W TLC Plates | Lipophilicity screening | Rapid compound ranking and profiling |
| Chiral Ligand Libraries | Stereocontrol in C–C bond formation | Asymmetric synthesis of sp3-rich centers |
| Electrocatalytic Systems | Sustainable redox mediation | C(sp3)–H functionalization under mild conditions |
| Photoredox Catalysts | Single-electron transfer activation | Radical-based C–C bond formation under visible light |
Computational and Informatics Approaches
The emergence of informacophore concepts represents a paradigm shift in developability optimization [117]. Unlike traditional pharmacophores based on human-defined heuristics, informacophores incorporate machine-learned representations of chemical structure combined with computed molecular descriptors to identify minimal structural requirements for biological activity while maintaining favorable physicochemical properties.
Machine learning algorithms can process ultra-large chemical datasets (e.g., Enamine's 65 billion make-on-demand compounds) to identify patterns beyond human perception [117]. These approaches enable:
- Prediction of optimal Fsp3 ranges for specific target classes
- Identification of lipophilicity sweet spots based on historical success rates
- Bioisosteric replacement suggestions that maintain potency while improving properties
- Retrosynthetic planning prioritizing routes that enhance molecular complexity
The integration of these computational approaches with experimental validation creates an iterative feedback loop that systematically addresses developability challenges while maintaining biological efficacy [117].
Addressing developability challenges through conscious reduction of lipophilicity and enhancement of Fsp3 requires integrated application of sophisticated synthetic methodology, robust analytical characterization, and predictive computational tools. By leveraging modern reaction mechanisms including molecular electrocatalysis, photoredox chemistry, and innovative C–C bond-forming reactions, medicinal chemists can deliberately design compounds with improved developmental trajectories. The ongoing evolution of reaction mechanisms in organic chemistry continues to provide transformative tools for constructing complex, sp3-rich molecular architectures with optimized physicochemical properties, ultimately enhancing the efficiency of drug discovery and the quality of resulting therapeutic agents.
Practical Guide to Troubleshooting Common Laboratory Reaction Failures
Within the broader thesis on reaction mechanisms in organic chemistry research, the inability to obtain a desired product is not a dead end but a critical diagnostic tool. Reaction failures provide invaluable insights into the subtle nuances of chemical mechanisms and the practical parameters that govern them. For researchers and drug development professionals, developing a systematic troubleshooting methodology is as essential as mastering synthesis itself. This guide provides a structured framework to diagnose and correct common laboratory reaction failures, emphasizing the mechanistic underpinnings that explain experimental observations. Moving beyond "voodoo" practices and into the realm of deliberate, hypothesis-driven investigation, this approach transforms failed experiments from setbacks into learning opportunities, accelerating the pace of research and development [118].
A Systematic Troubleshooting Methodology
Effective troubleshooting is a logical, step-by-step process designed to efficiently isolate the root cause of a reaction failure. The following workflow provides a structured approach for diagnosis. The corresponding diagram visualizes this systematic methodology.
Diagram 1: A systematic workflow for diagnosing reaction failures.
The process begins with fundamental verification before progressing to complex mechanistic problem-solving. Each stage is critical for an efficient diagnosis.
- Verify the Reaction Setup: Before postulating complex mechanistic failures, confirm the basics. Check that starting materials and reagents are correct, pure, and have been stored properly. Ensure glassware is clean and dry, and that the reaction was set up under the correct atmosphere (e.g., inert N₂ or Ar) if moisture- or oxygen-sensitive [118].
- Analyze the Crude Mixture: Use thin-layer chromatography (TLC), LC-MS, NMR spectroscopy, or other analytical techniques to probe the reaction mixture. The goal is to determine if the starting material is consumed, if new spots/peaks are present, and if the desired product is formed, even in low yield [118].
- Identify the Symptom: Based on the analysis, categorize the failure into a specific symptom, such as no reaction, low yield, formation of a side product, or reaction stall.
- Formulate a Mechanistic Hypothesis: This is the core of troubleshooting. Based on the observed symptom and your knowledge of the reaction mechanism, propose a plausible chemical explanation.
- Design and Execute a Diagnostic Experiment: Test your hypothesis with a targeted experiment. This should be a small-scale, quick test designed to prove or disprove the proposed cause.
- Interpret Data and Refine Approach: Analyze the results of your diagnostic experiment. If the hypothesis is confirmed, apply the corrective action. If not, return to Step 4 and formulate a new hypothesis based on the new data.
Common Failure Scenarios and Diagnostic Strategies
This section details specific failure modes, their potential mechanistic causes, and targeted diagnostic experiments. The following table summarizes the core quantitative data and solutions for these common scenarios.
Table 1: Common Reaction Failure Scenarios and Diagnostic Solutions
| Failure Symptom | Key Quantitative Data & Observation | Proposed Mechanistic Cause(s) | Diagnostic Experiment & Solution |
|---|---|---|---|
| No Consumption of Starting Material | Starting material recovery >90% by TLC/NMR. | Reagent Degradation: Titration shows t-BuLi concentration <80% of stated [118].Incorrect Reaction Conditions: Temperature too low for activation energy.Inhibitory Impurities: Trace water or oxygen quenches reactive species. | Diagnostic: Titrate organometallic reagents [118]. Run reaction with activated molecular sieves.Solution: Use fresh reagents. Apply elevated temperature. Scrupulously exclude air/moisture. |
| Low Yield of Desired Product | Yield <40%; desired product identified by LC-MS. | Competitive Side Reactions: Elimination outcompetes substitution (e.g., E2 vs SN2) [119].Incomplete Reaction: Equilibrium favors starting materials.Product Degradation: Product is acid/base-sensitive or unstable at reaction T. | Diagnostic: Analyze crude mixture for side products. Monitor reaction progress over time.Solution: Modify electronic/steric environment (change base/nucleophile). Drive equilibrium (remove water, use excess reagent). Quench reaction promptly under mild conditions. |
| Formation of Major Side Product | Desired product <20%; one major side product isolated. | Regioselectivity Issue: Functionalization at less hindered position is kinetically favored but leads to wrong isomer.Over-reaction: Product is more reactive than starting material (e.g., poly-halogenation).Rearrangement: Reaction intermediate (e.g., carbocation) undergoes hydride or alkyl shift. | Diagnostic: Isolate and characterize side product (NMR, IR, MS).Solution: Change catalyst/conditions to favor thermodynamic product. Use protecting groups. Employ stoichiometric control (e.g., 1.0 eq oxidant). |
| Reaction Stall | ~50% SM remains by TLC; no further change. | Catalyst Deactivation: Catalyst is poisoned by an impurity.Reagent Depletion: Limiting reagent is consumed.Temperature Sensitivity: Enzyme or catalyst is inactive at current T. | Diagnostic: Add fresh catalyst/reagent to stalled mixture. Monitor reaction temperature with internal probe.Solution: Recharge with fresh catalyst. Use excess of one reagent. Optimize temperature profile. |
The "No Reaction" Scenario
When the starting material remains entirely unconsumed, the issue often lies in the failure to generate the active catalytic or reactive species.
- Mechanistic Insight: Many reactions proceed through a high-energy, activated complex. If the energy barrier for its formation is not met, the reaction will not initiate. This is common in organometallic catalysis and reactions involving anhydride formation.
- Experimental Protocol for Titration: To diagnose degraded reagents, titrate organolithium reagents against a known concentration of a substrate like menthol in THF, using 1,10-phenanthroline as a colorimetric indicator. The endpoint is a persistent orange-red color, allowing calculation of the exact molarity [118].
- Corrective Protocol: If reagents are degraded, replace them with fresh or properly stored stocks. For new reactions, ensure the reaction temperature is sufficient. A simple test is to run a small-scale reaction and gradually increase the temperature while monitoring by TLC.
The Low Yield Conundrum
The formation of some product, but in disappointingly low yield, indicates the reaction is proceeding but is inefficient.
- Mechanistic Insight: Low yields often result from competitive reaction pathways. For instance, a base can deprotonate a substrate to form an anion for desired reactivity, but it can also act as a nucleophile to cause elimination, or as a base to promote hydrolysis. The outcome depends on the delicate balance of sterics, electronics, and conditions [119].
- Diagnostic Protocol: Analyze the crude reaction mixture thoroughly. Use preparative TLC or a small-scale flash column to isolate the major components of the mixture. Characterizing the side products provides direct clues about the competing pathway. For example, finding an elimination alkene points to a basic impurity or too high a temperature.
- Corrective Protocol: Employ the strategy of "reaction mapping" to find an alternative route. If a direct conversion of an alkane to a thiol fails, a two-step "connecting flight" via an alkyl halide might be successful (e.g., alkane → alkyl chloride → thiol) [119]. This involves viewing functional groups as hubs and reactions as connecting flights to plan multi-step syntheses.
Advanced Diagnostic and Analytical Techniques
In-Reaction Monitoring
Relying solely on an endpoint analysis is insufficient for complex reactions.
- Protocol for TLC Monitoring:
- Spotting: Use a micro-capillary spotter to withdraw a ~10 µL aliquot from the reaction flask.
- Quenching: Dilute the aliquot into a few drops of a benign, volatile solvent (e.g., ethyl acetate or DCM) in a small vial to quench the reaction.
- Visualization: Spot the quenched solution on a TLC plate alongside authentic samples of the starting material and desired product. Develop the plate in an appropriate mobile phase and visualize using UV light and/or an appropriate stain (e.g., KMnO₄, vanillin).
- Data Interpretation: Track the disappearance of the starting material (lower Rf) and the appearance of the product (often, but not always, a higher Rf). The presence of multiple new spots indicates side reactions.
The Critical Role of Work-Up and Purification
A successful reaction can be ruined during work-up and purification. A proper work-up is designed to isolate the crude product from the reaction mixture, terminating the reaction and removing solvents, catalysts, and soluble by-products [120].
- Work-Up Protocol (Liquid-Liquid Extraction):
- Quenching: Once the reaction is complete (by TLC), carefully add the mixture to a separatory funnel containing water or a quenching solution (e.g., saturated aqueous NH₄Cl for Grignard reactions).
- Separation: Add an immiscible organic solvent (e.g., diethyl ether or ethyl acetate), shake with frequent venting, and allow the layers to separate completely.
- Extraction: Drain and save the organic layer. Re-extract the aqueous layer with fresh organic solvent (2-3 times).
- Drying: Combine the organic extracts and dry them over a anhydrous inorganic salt (e.g., MgSO₄ or Na₂SO₄).
- Concentration: Filter off the drying agent and concentrate the filtrate under reduced pressure using a rotary evaporator to obtain the crude product [120].
- Purification Protocol (Flash Column Chromatography): This is a primary purification technique. The crude product is loaded onto a column packed with a stationary phase (e.g., silica gel). A mobile phase (solvent or mixture) is passed through the column under positive pressure, eluting different compounds at different rates based on their polarity.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table catalogues key reagents and materials critical for both executing and troubleshooting synthetic reactions.
Table 2: Essential Reagents and Materials for Reaction Troubleshooting
| Item | Function & Application in Troubleshooting |
|---|---|
| Molecular Sieves (3Å, 4Å) | Used to remove residual water from reaction mixtures. Essential for troubleshooting water-sensitive reactions (e.g., Grignard formation, organometallic couplings) [118]. |
| TLC Plates & Visualization Stains | For monitoring reaction progress. Critical for diagnosing "no reaction," "stall," or "side product" scenarios by providing a rapid snapshot of reaction composition [118]. |
| Triphenylphosphine Oxide | A common, crystalline by-product used as a visual marker to gauge solvent polarity during flash column chromatography method development. |
| Deuterated Solvents (CDCl₃, DMSO-d6) | For NMR analysis of crude reaction mixtures and purified compounds. Essential for identifying and characterizing unknown side products and confirming product structure. |
| SCX (Strong Cation Exchange) Cartridges | Used in work-up to remove basic impurities by binding them to the solid phase, simplifying the purification of the desired neutral product. |
Visualizing Synthetic Strategy with Reaction Maps
Understanding how functional groups interconvert is key to planning synthesis and troubleshooting. A reaction map visualizes this knowledge, showing possible synthetic pathways. The diagram below illustrates a simplified map for alkane and alkyl halide functionalization.
Diagram 2: A reaction map showing key transformations from alkanes and alkyl halides.
This map demonstrates that an alkyl halide acts as a central "hub," enabling diverse synthetic transformations [119]. When a direct synthetic route fails, this map-based thinking allows a researcher to plan a multi-step "connecting flight" to the target. For example, converting an alkane directly to a thiol may be impossible, but a two-step route via an alkyl halide is often feasible [119].
Troubleshooting failed reactions is a fundamental skill in organic chemistry research. By adopting a systematic, hypothesis-driven approach—from verifying basic setup and analyzing the crude mixture to formulating mechanistic hypotheses and designing diagnostic experiments—researchers can efficiently diagnose and overcome synthetic challenges. This guide underscores that understanding reaction mechanisms is not merely an academic exercise but the most powerful tool in the practical chemist's arsenal. Integrating these structured troubleshooting strategies, analytical techniques, and the conceptual framework of reaction maps will significantly enhance research productivity and success in drug development and complex molecule synthesis.
Validating and Comparing Mechanistic Pathways for Predictive Accuracy
In the field of organic chemistry research, particularly within pharmaceutical development, the validation of reaction mechanisms represents a critical bridge between theoretical models and practical application. Mechanism validation is defined as the process of establishing rigorous, documented evidence that provides a high degree of assurance that a proposed reaction mechanism accurately represents the actual molecular steps occurring in a chemical transformation [121]. This process moves beyond merely observing reaction outcomes to systematically demonstrating causality within complex molecular systems [122].
The philosophical foundation for mechanistic validation lies in scientific falsifiability, where good mechanistic theories "forbid certain things to happen" and become more robust when they survive genuine attempts at refutation [122]. Within organic chemistry, this translates to proposing detailed molecular pathways—including bond-breaking and bond-forming processes, intermediates, and transition states—and then designing critical experiments that can potentially disprove these pathways [123]. The shift from phenomenological observation (simply noting that a reaction occurs) to mode-of-action understanding (describing key events) and finally to mechanistic validation (providing a detailed molecular description) represents an evolution in how organic chemists approach reaction understanding [122].
For researchers and drug development professionals, rigorous mechanism validation is not merely an academic exercise but a practical necessity. Validated mechanisms enable predictive reaction design, optimization of synthetic routes, and troubleshooting of scale-up processes in pharmaceutical manufacturing. Furthermore, regulatory agencies increasingly expect scientific evidence demonstrating understanding of critical process parameters and their relationship to product quality attributes, particularly for complex reactions involved in active pharmaceutical ingredient synthesis [121].
Theoretical Frameworks for Establishing Causality
Bradford Hill Criteria Adaptation for Chemistry
The validation of reaction mechanisms requires frameworks for establishing causality in complex systems. While originally developed for epidemiological studies, the Bradford Hill criteria provide a valuable adapted framework for assessing mechanistic causality in organic chemistry [122]. This approach offers multiple types of evidence that collectively support a mechanistic hypothesis without requiring unrealistically simple linear relationships:
- Strength of the Correlation: The magnitude of the effect between proposed cause and observed outcome
- Consistency: Reproducibility of the mechanistic pathway across different experimental conditions
- Specificity: Demonstration that specific molecular features yield predictable and specific outcomes
- Temporality: Experimental confirmation that proposed intermediates occur before products in the reaction timeline
- Biological Gradient (adapted as "Reaction Gradient"): Observation of a dose-response relationship where changes in reactant concentration or reaction conditions produce proportional changes in outcomes
- Plausibility: Consistency of the proposed mechanism with established chemical principles and quantum mechanical calculations
- Coherence: Agreement between the proposed mechanism and multiple analytical techniques
- Experiment: Evidence from controlled intervention studies specifically designed to test the mechanism
- Analogy: Comparison with previously validated mechanisms in similar chemical systems
This multi-factorial approach acknowledges the complexity of chemical systems while providing a structured methodology for building confidence in mechanistic proposals. Unlike the more rigid Koch-Dale postulates, which assume linear causality, the Bradford Hill framework accommodates the networked reality of molecular interactions and competing pathways [122].
Distinguishing Between Mode of Action and Mechanism
A crucial distinction in mechanistic validation lies between mode of action and mechanism of action. As defined in regulatory toxicology and applicable to organic chemistry:
- Mode of Action: "The description of key events and processes, starting with interaction of an agent with the cell through functional and anatomical changes, resulting in cancer or other health endpoints" [122]
- Mechanism of Action: "The detailed molecular description of key events in the induction of cancer or other health endpoints" [122]
Translated to organic chemistry context, mode of action describes the observable key events in a reaction pathway (formation of detectable intermediates, stereochemical outcomes, kinetic profiles), while mechanism of action provides the detailed molecular description including transition state structures, orbital interactions, and precise bond-forming/breaking sequences. Complete mechanistic validation aims to elevate understanding from mode of action to mechanism of action wherever possible.
Experimental Methodologies for Mechanism Validation
Analytical Techniques for Mechanistic Studies
Validating reaction mechanisms requires the application of multiple complementary analytical techniques to gather diverse lines of evidence. The table below summarizes key methodologies and their specific applications in mechanistic studies:
Table 1: Analytical Techniques for Mechanistic Validation in Organic Chemistry
| Technique | Application in Mechanism Validation | Key Experimental Parameters |
|---|---|---|
| Nuclear Magnetic Resonance (NMR) Spectroscopy | Detection and characterization of reactive intermediates, determination of stereochemistry | Variable temperature studies, kinetic profiling, trapping experiments |
| Infrared (IR) Spectroscopy | Monitoring functional group transformations, identifying carbonyl intermediates | Time-resolved studies, monitoring bond vibration changes |
| Mass Spectrometry (MS) | Identifying molecular ions of intermediates, isotopic labeling studies | High-resolution accuracy, tandem MS for fragmentation pathways |
| Ultraviolet-Visible (UV-Vis) Spectroscopy | Monitoring conjugated systems, reaction kinetics | Stopped-flow techniques for rapid kinetics |
| X-ray Crystallography | Determining precise molecular geometry of intermediates | Single crystal growth of stable intermediates or analogs |
| Computational Chemistry | Modeling transition states, calculating energy profiles, predicting spectroscopic properties | Density functional theory (DFT), ab initio methods, solvation models |
The integration of multiple techniques provides orthogonal validation, where consistent results across different methodologies significantly strengthen mechanistic proposals [123]. For example, an intermediate detected by NMR should have a corresponding molecular ion in MS data and calculated properties matching computational predictions.
Kinetic Analysis Techniques
Kinetic studies provide critical evidence for distinguishing between potential mechanisms by quantifying reaction rates and their dependence on reaction conditions. The table below outlines essential kinetic approaches:
Table 2: Kinetic Methods for Mechanistic Validation
| Method | Mechanistic Information | Experimental Protocol |
|---|---|---|
| Initial Rates Method | Determination of reaction order with respect to each reactant | Vary one reactant concentration while keeping others in large excess; measure initial rate |
| Integrated Rate Laws | Verification of proposed molecularity | Monitor concentration vs. time; fit to zero, first, or second-order models |
| Eyring Analysis | Determination of activation parameters (ΔH‡, ΔS‡) | Measure rate constants at multiple temperatures; plot ln(k/T) vs. 1/T |
| Isotope Effects | Identification of bond-breaking in rate-determining step | Substitute with deuterium or other isotopes; measure kinetic isotope effect (KIE) |
| Competition Kinetics | Relative reactivity of different substrates | React two substrates simultaneously with limiting reagent; analyze product ratio |
| Crossover Experiments | Distinguishing between intramolecular vs. intermolecular pathways | Use structurally distinguishable but chemically similar reactants |
These kinetic methods enable researchers to move beyond stoichiometric observations to dynamic understanding of reaction pathways, providing critical evidence for or against proposed mechanisms.
Experimental Design for Mechanistic Studies
Effective mechanistic validation employs strategic experimental designs that specifically test predictions of proposed mechanisms:
- Critical Intermediate Trapping: Introduction of chemical traps (e.g., nucleophiles, radicals, dienophiles) to intercept proposed reactive intermediates
- Stereochemical Probes: Use of stereodefined substrates to probe stereoelectronic requirements and conservation or inversion of configuration
- Cross-Over Experiments: Distinguishing between intra- and intermolecular pathways using structurally distinguishable reagents
- Isotopic Labeling: Strategic placement of isotopes (²H, ¹³C, ¹⁸O) to track atom movement through reaction pathways
- Linear Free Energy Relationships: Correlation of reaction rates with substrate electronic or steric parameters (Hammett, Taft plots)
- Solvent Effects: Systematic variation of solvent properties to probe charge development in transition states
Each experimental approach should be designed not merely to confirm the proposed mechanism but to genuinely attempt falsification by testing non-obvious predictions [122].
Data Presentation and Statistical Correlation
Quantitative Correlation of Experimental and Computational Data
Robust mechanistic validation requires quantitative correlation between experimental observations and computational predictions. The table below illustrates key parameters for comparison:
Table 3: Correlation Metrics for Mechanistic Validation
| Parameter | Experimental Measurement | Computational Prediction | Validation Criteria |
|---|---|---|---|
| Activation Energy | From Arrhenius or Eyring plot (kJ/mol) | Energy difference between reactant and transition state | Absolute error < 10-15 kJ/mol |
| Reaction Energy | Calorimetry, equilibrium constant (kJ/mol) | Energy difference between reactants and products | Absolute error < 15-20 kJ/mol |
| Bond Lengths | X-ray crystallography (Å) | Optimized geometry (Å) | R² > 0.95 for correlation |
| Vibrational Frequencies | IR/Raman spectroscopy (cm⁻¹) | Calculated frequencies (cm⁻¹) | Scaling factor ~0.96-0.98 |
| Chemical Shifts | NMR spectroscopy (ppm) | GIAO calculations (ppm) | R² > 0.95 for correlation |
| Isotope Effects | Kinetic measurements (KIE) | Calculated frequencies (KIE) | Agreement within 10-20% |
Statistical measures of correlation quality should be reported, including correlation coefficients (R²), confidence intervals, and measures of predictive accuracy [121]. Modern approaches increasingly use machine learning algorithms to identify complex patterns in multidimensional mechanistic data that might escape conventional analysis.
Statistical Approach to Validation
The integration of Six Sigma principles into mechanistic validation brings statistical rigor through [121]:
- Sample Size Determination: Based on statistical power analysis to detect meaningful effects
- Confidence Intervals: Quantifying uncertainty in measured parameters
- Hypothesis Testing: Formal statistical tests to confirm or reject mechanistic predictions
- Control Charts: Monitoring analytical method performance over time
- Capability Analysis: Quantifying how well a mechanistic model predicts experimental outcomes
This statistical approach transforms mechanistic validation from qualitative agreement to quantitative assessment of predictive capability.
Research Reagent Solutions for Mechanistic Studies
Table 4: Essential Research Reagents for Mechanistic Investigations
| Reagent Category | Specific Examples | Function in Mechanistic Studies |
|---|---|---|
| Spin Traps | DMPO, PBN, TEMPO | Detection and characterization of radical intermediates |
| Isotopically Labeled Compounds | ¹³C-labeled substrates, D₂O, ¹⁸O₂ | Tracing atom movement through reaction pathways |
| Chemical Quenching Agents | Trialkylphosphites, hydrazines | Trapping reactive oxygen species or other transient intermediates |
| Coordinating Solvents | Hexamethylphosphoramide, crown ethers | Probing cation-π interactions or coordination effects |
| Radical Initiators | AIBN, benzoyl peroxide | Generating radical species under controlled conditions |
| Inhibitors/Scaverngers | BHT, galvinoxyl, superoxide dismutase | Testing for specific mechanistic pathways by inhibition |
| Lewis Acids/Bases | BF₃·Et₂O, DBU | Probing acid/base catalysis in mechanistic pathways |
These specialized reagents enable researchers to design critical experiments that test specific aspects of proposed mechanisms, particularly for distinguishing between alternative pathways.
Visualization of Mechanistic Validation Workflows
Mechanistic Validation Workflow
Experimental Correlation Methodology
Experimental Correlation Pathway
The validation of reaction mechanisms through correlation with experimental data represents a cornerstone of modern organic chemistry research, particularly in pharmaceutical development where understanding molecular transformations directly impacts product quality and patient safety. By employing the structured frameworks, experimental methodologies, and statistical approaches outlined in this guide, researchers can move beyond phenomenological observations to establish causally verified mechanistic understanding. The integration of multi-modal analytical data with computational predictions, assessed through adapted Bradford Hill criteria, provides a robust foundation for mechanistic claims that withstand scientific scrutiny and support regulatory requirements. As reaction mechanisms become increasingly complex in modern synthetic methodology, these validation principles will continue to ensure that theoretical models remain grounded in experimental reality.
Comparative Analysis of Different Mechanistic Pathways for the Same Transformation
In organic chemistry, a single chemical transformation can often proceed via multiple, distinct mechanistic pathways. The ability to predict, analyze, and control which pathway dominates is fundamental to synthetic efficiency, selectivity, and ultimately, the success of research in fields like drug development. Understanding these pathways is not merely an academic exercise; it allows scientists to rationally design reactions, minimize byproducts, and optimize conditions for industrial and pharmaceutical applications [124]. This guide provides a structured, data-driven framework for the comparative analysis of competing mechanistic pathways, contextualized within modern computational and experimental practices. The analysis of these pathways forms a core thesis in contemporary organic chemistry research: that mechanistic understanding, rather than heuristic rules, should guide synthetic design.
Theoretical Framework for Mechanistic Differentiation
Key Decision Factors in Pathway Selection
The journey from reactants to products is governed by a set of controllable and inherent chemical factors that determine the operative mechanism. For researchers, a systematic investigation of these factors is the first step in any mechanistic study.
Table 1: Key Factors Influencing Mechanistic Pathway Selection
| Factor Category | Specific Parameter | Impact on Mechanism |
|---|---|---|
| Electronic Effects | Substituent Electronic Nature (EDG/EWG) | Stabilizes/destabilizes charged intermediates; influences nucleophilicity/electrophilicity [124]. |
| Conjugation and Aromaticity | Can enable resonance stabilization of intermediates, favoring pathways like SN1 or E1 [124]. | |
| Steric Effects | Substrate Steric Hindrance | Highly hindered substrates favor unimolecular pathways (SN1, E1) over bimolecular ones (SN2, E2) [124]. |
| Base/Nucleophile Size | Large, bulky nucleophiles are better bases than nucleophiles, favoring elimination over substitution. | |
| Reaction Conditions | Reaction Temperature | Higher temperatures typically increase the prevalence of elimination over substitution and favor unimolecular reactions [125]. |
| Solvent Polarity and Proticity | Polar protic solvents stabilize ions, favoring SN1/E1; polar aprotic solvents enhance nucleophile strength, favoring SN2 [126]. | |
| pH and Catalyst | Acid or base catalysis can open entirely different mechanistic avenues, such as in acid-catalyzed naphthalene transformations [127]. |
A Framework for Mechanistic Assignment
The definitive assignment of a reaction mechanism requires a multi-faceted approach, moving beyond simple product identification.
- Experimental Kinetic Data: Determining the reaction order with respect to the substrate and nucleophile/base is a classical but powerful tool. For instance, a first-order rate law suggests a unimolecular mechanism (SN1/E1), while a second-order law suggests a bimolecular one (SN2/E2).
- Stereochemical Analysis: The inversion of configuration at a stereocenter is a hallmark of the SN2 mechanism. In contrast, racemization is characteristic of the SN1 pathway, which proceeds through a planar, achiral carbocation intermediate.
- Intermediate Trapping and Isotope Labeling: The use of spectroscopic methods or chemical traps to detect transient intermediates (e.g., carbocations) provides direct evidence. Isotopic labeling, such as with deuterium, can help track atom movements and kinetic isotope effects [127].
- Computational Chemistry: Density Functional Theory (DFT) calculations can model reaction pathways, locate transition states, and calculate activation barriers, providing a theoretical energy landscape that supports or refutes a proposed mechanism [127].
Case Study: Nucleophilic Substitution at a Saturated Carbon
The competition between SN1, SN2, SN1', and SN2' mechanisms for nucleophilic substitution serves as a paradigmatic example for comparative mechanistic analysis.
Pathway Descriptions and Energy Profiles
The following diagram illustrates the logical decision process for identifying the most probable mechanistic pathway based on substrate and conditions, integrating concepts from the analysis of competing reactions.
Comparative Analysis of Substitution Pathways
Table 2: Quantitative Comparison of Nucleophilic Substitution Mechanisms
| Mechanism | Molecularity | Rate Law | Stereochemical Outcome | Key Intermediate | Susceptibility to Rearrangement |
|---|---|---|---|---|---|
| SN2 | Bimolecular | k[Substrate][Nu] | Inversion of configuration | None (Concerted) | No |
| SN1 | Unimolecular | k[Substrate] | Racemization | Carbocation | Yes [124] |
| SN2' | Bimolecular | k[Substrate][Nu] | Specific stereochemistry based on allylic system | None (Concerted) | No |
| SN1' | Unimolecular | k[Substrate] | Mixture of stereoisomers | Allylic Carbocation | Yes |
Advanced Case Study: Electrophilic Aromatic Substitution vs. Addition
The reaction of an alkene-substituted aromatic ring (like styrene) with an electrophile (E+) can diverge onto two fundamentally different pathways: electrophilic aromatic substitution (SEAr) or electrophilic addition to the alkene (AdE). The following workflow models the reasoning process to predict the major product.
Experimental Protocols for Mechanistic Elucidation
Protocol A: Kinetic Order Determination
Objective: To distinguish between unimolecular and bimolecular mechanisms by measuring the reaction rate as a function of reactant concentrations.
- Reaction Setup: Prepare a series of reaction vials under an inert atmosphere. The temperature must be controlled precisely using a thermostated bath [125].
- Variation of Concentrations: For a reaction between substrate S and nucleophile N, perform two sets of experiments:
- Set 1: Keep [N] in large excess (pseudo-first-order conditions) and vary [S].
- Set 2: Keep [S] in large excess and vary [N].
- Sampling and Analysis: At regular time intervals, withdraw aliquots from the reaction mixture. Quench the reaction instantly and analyze the concentration of a product or the remaining substrate using a suitable technique (e.g., GC, HPLC, NMR).
- Data Analysis: Plot the concentration data to determine the rate constant for each run. The reaction order is determined by the dependence of the observed rate constant on the concentration of the varied species.
Protocol B: Crossover Experiment for Intermolecularity
Objective: To detect whether a reaction proceeds through an intermolecular intermediate, which is strong evidence for a dissociative (e.g., SN1) mechanism.
- Design of Crossover Substrates: Synthesize two structurally similar substrates that are distinguishable by an analytical tag (e.g., isotopic label, distinct substituents like R and R').
- Execution: Mix the two substrates (e.g., R-X and R'-X) and subject them to the reaction conditions in the same vessel.
- Product Analysis: Analyze the products using mass spectrometry or NMR. The detection of "crossover" products (e.g., R-Y and R'-Y from R-X and R'-X is straightforward), but also R-R' or mixed species, indicates that free, intermolecular species (e.g., carbocations R+ and R'+) were generated and combined randomly.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Mechanistic Studies
| Reagent / Material | Function in Mechanistic Analysis |
|---|---|
| Deuterated Solvents (e.g., CDCl₃, D₂O) | Essential for NMR spectroscopy to monitor reaction progress, identify products, and track deuterium incorporation in labeling studies [127]. |
| Isotopically Labeled Compounds (e.g., ¹³C, ²H) | Act as "spies" to track atom fate through a reaction mechanism, enabling the determination of kinetic isotope effects and validation of proposed pathways [127]. |
| Radical Initiators (e.g., AIBN) & Inhibitors (e.g., BHT) | Used to probe for radical mechanisms. An initiator should accelerate a radical chain reaction, while an inhibitor will suppress it. |
| Strong Lewis Acids (e.g., AlCl₃, BF₃) | Used to catalyze reactions involving carbocation intermediates, such as Friedel-Crafts alkylation, and to study their formation and stability [127]. |
| Scavenger Reagents | Compounds designed to trap and characterize reactive intermediates (e.g., alkenes to trap carbocations as new adducts). |
| Computational Chemistry Software | Tools for DFT calculations (e.g., Gaussian, ORCA) to model potential energy surfaces and identify transition states, providing theoretical support for a mechanism [127]. |
The systematic comparative analysis of mechanistic pathways is a cornerstone of modern organic chemistry. By integrating classical experimental techniques—such as kinetic studies and stereochemical analysis—with advanced tools like isotopic labeling and computational modeling, researchers can move beyond simple product prediction to a profound understanding of molecular behavior. This deep mechanistic insight is indispensable for driving innovation in complex synthetic endeavors, including the efficient and selective synthesis of active pharmaceutical ingredients (APIs) and novel materials. As datasets like mech-USPTO-31K [124] and benchmarks like oMeBench [128] continue to grow, they will further empower data-driven and AI-assisted discovery, solidifying the role of mechanistic reasoning as the fundamental algorithm for reaction prediction and design.
Using Ignition Delay Times and Species Concentration Profiles for Combustion Model Validation
The development of reliable chemical kinetic models is a cornerstone of modern combustion research, with direct implications for engine design, emission reduction, and the utilization of alternative fuels. Within the broader context of organic chemistry research, particularly in understanding complex reaction mechanisms, the validation of these models against precise experimental data is paramount. This whitepaper provides an in-depth technical guide on employing two critical datasets—ignition delay times (IDTs) and species concentration profiles—for rigorous combustion model validation. IDT measurements offer global validation targets that characterize overall fuel reactivity, while species profiles provide fundamental, time-resolved insights into the intricate chemical pathways governing fuel decomposition and oxidation. The synergistic use of these data types enables researchers to develop and refine kinetic models with predictive capabilities across a wide range of temperatures, pressures, and chemical environments, thereby accelerating the development of cleaner combustion technologies and novel chemical processes.
Fundamental Combustion Validation Targets
Ignition Delay Time (IDT)
Ignition Delay Time (IDT) is a fundamental property that characterizes the global reactivity of a fuel-oxidizer mixture. It is universally defined as the time interval between a combustible mixture receiving a thermal stimulus (e.g., rapid compression in a Rapid Compression Machine - RCM, or heating by a reflected shock wave in a shock tube) and the onset of a violent chemical reaction, typically indicated by a rapid pressure rise or light emission [129] [130]. IDT data serves as a primary, global validation target for chemical kinetic mechanisms, testing the model's ability to accurately predict the overall ignition behavior under a wide range of conditions.
The experimental apparatus used to measure IDT is selected based on the target temperature and time scales. Shock Tubes (ST) are ideal for high-temperature IDTs ranging from microseconds to a few milliseconds, achieved almost instantaneously through shock wave compression [129] [130]. Conversely, Rapid Compression Machines (RCM) achieve low to intermediate temperatures and high pressures through a fast piston compression, and are effective for measuring IDTs from 10 to hundreds of milliseconds [130] [131]. The thermal conditions in an RCM are less ideal than in a shock tube due to finite compression times and significant heat losses, leading to potential deviations in measured IDTs between different facilities. Standardization methods, such as the inverse Livengood-Wu integral, have been proposed to correlate these measurements to ideal, adiabatic constant-volume conditions [130].
Species Concentration Profiles
While IDT provides a global measure of reactivity, species concentration profiles deliver a far more detailed, time-resolved validation target. These profiles track the evolution of key chemical species—reactants, intermediates, and products—during the combustion process, offering a direct window into the underlying chemical reaction pathways.
These profiles can be measured in various experimental setups, including:
- Flow Reactors: These provide data on species evolution under well-controlled temperature and pressure conditions, often used for pyrolysis and oxidation studies [131] [132]. For instance, the pyrolysis of n-decane has been studied in flow reactors to identify major decomposition pathways [131].
- Jet-Stirred Reactors (JSR): JSRs operate under perfect mixing conditions, providing concentration data for stable and unstable species at a fixed residence time while temperature is varied [133].
- Shock Tubes: Advanced diagnostic techniques in shock tubes can also be used to record time-histories of specific species during the ignition process [129].
The temperature profile within a reactor is critical, as a temperature rise can accelerate the primary reaction but also trigger undesirable side reactions [132]. Validating a model against species profiles ensures that not only the global ignition behavior is correct, but also the intricate network of elementary reactions is accurately captured.
Experimental Methodologies and Protocols
Protocol for Ignition Delay Time Measurement in a Shock Tube
The high-temperature IDTs for ammonia/dimethoxymethane (NH3/DMM) mixtures, as detailed by Dai et al. [129], serve as an exemplary protocol for shock tube operation:
- Facility Preparation: Utilize a stainless-steel shock tube separated by polyester terephthalate (PET) diaphragms into an 8 m driven section and a 4 m driver section.
- Mixture Preparation: Prepare test gas mixtures in a stainless-steel tank using the partial pressure method. Ensure mixture homogeneity by letting the gases mix for at least 24 hours before use. Verify composition via gas chromatography.
- Instrumentation: Mount multiple piezoelectric pressure transducers (e.g., PCB 111A24) at fixed intervals along the driven section to measure incident shock velocity. Monitor pressure time-histories near the endwall using a high-frequency response pressure transducer (e.g., Kistler 603B1).
- Ignition Event Determination: Conduct experiments behind reflected shock waves. Define the IDT as the time interval between the arrival of the reflected shock wave at the endwall and the subsequent rapid pressure rise associated with ignition, as identified by the maximum slope in the pressure history (dp/dt)max.
- Condition Definition: Calculate the temperature and pressure behind the reflected shock wave (T5 and P5) using standard shock relations and the measured incident shock velocity.
This methodology has been successfully applied to measure IDTs of NH3/DMM mixtures at DMM blending ratios of 5-50%, pressures of 1 and 10 bar, an equivalence ratio of 0.5, and temperatures between 1193 and 1852 K [129].
Protocol for Species Concentration Profiling in a Flow Reactor
The work of Zeng et al. on n-decane pyrolysis, cited in Liu et al. [131], outlines a standard approach for obtaining species profiles:
- Reactor Setup: Employ a flow reactor capable of operating over a wide range of pressures (e.g., 5 to 760 Torr) and temperatures (e.g., 780–1500 K).
- Fuel Introduction and Heating: Introduce the fuel vapor, typically carried by an inert gas like argon or helium, into the heated reactor zone.
- Sample Extraction: Use a movable sampling probe to extract gas samples from along the length of the reactor (corresponding to different reaction times/temperatures).
- Species Analysis: Analyze the gas samples using gas chromatography (GC) or mass spectrometry (GC/MS) to quantify the concentrations of reactants, stable intermediates, and final products.
- Data Correlation: Correlate the position in the reactor with residence time to construct species concentration profiles as a function of reaction progress.
This protocol identified that n-decane is primarily consumed by C-C bond dissociation and H-atom abstraction by methyl radicals, with measured 1-olefin concentrations being highly sensitive to these decomposition pathways [131].
Data Integration Workflow
The following diagram illustrates the integrated workflow for conducting experiments and using the data for model validation:
Data Presentation and Kinetic Analysis
Table 1: Summary of key experimental data for combustion model validation.
| Fuel / Mixture | Experimental Target | Conditions | Apparatus | Key Observations for Validation |
|---|---|---|---|---|
| NH₃ / Dimethoxymethane (DMM) [129] | Ignition Delay Time (IDT) | ( P = 1, 10 \, \text{bar} ), ( \phi = 0.5 ), ( T = 1193-1852 \, \text{K} ) | Shock Tube | High-pressure data constrains key cross-reactions (e.g., DMM + NH₂). |
| Methane / n-Decane [131] | IDT & Species Profiles | ( P = 30 \, \text{bar} ), ( \phi = 0.5, 1.0, 2.0 ), ( T = 629-1494 \, \text{K} ) | Shock Tube & RCM | Tests model from low-T (RCM) to high-T (ST) for dual-fuel application. |
| n-Decane [131] | Species Profiles (Pyrolysis) | ( P = 5-760 \, \text{Torr} ), ( T = 780-1500 \, \text{K} ) | Flow Reactor | Sensitive to C-C bond scission and H-abstraction by CH₃. |
| Ethanol [133] | IDT, Laminar Flame Speed, Species | Various pressures and temperatures | ST, RCM, Flames, JSR | Comprehensive validation for a core biofuel mechanism. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key reagents, materials, and equipment used in combustion experiments.
| Item | Function / Relevance | Example from Literature |
|---|---|---|
| Shock Tube | Provides high-temperature, high-pressure conditions for measuring short IDTs. | Used for NH₃/DMM IDTs at T > 1193 K [129]. |
| Rapid Compression Machine (RCM) | Provides low-to-intermediate temperature, high-pressure conditions for measuring longer IDTs. | Used for methane/n-decane IDTs at T as low as 629 K [131]. |
| Flow Reactor / Jet-Stirred Reactor (JSR) | Provides species concentration data for pyrolysis and oxidation kinetics. | Used for n-decane pyrolysis and ethanol oxidation studies [131] [133]. |
| High-Purity Fuel Gases & Vapors | Ensure reproducible experiments free from impurities that could alter chemistry. | NH₃, CH₄, DMM vapor, n-decane vapor [129] [131]. |
| Piezoelectric Pressure Transducers | Critical for tracking pressure history and determining IDT in ST and RCM. | PCB 111A24 and Kistler 603B1 sensors used [129] [130]. |
| Gas Chromatograph / Mass Spectrometer (GC/MS) | Essential for quantifying species concentration profiles from reactors. | Used to analyze products from n-decane pyrolysis [131]. |
Advanced Modeling and Theoretical Frameworks
Mechanism Development and Analysis
The construction of a robust kinetic model is a sophisticated process. The hierarchical approach remains a standard methodology, where a core mechanism for small molecules (C0-C3) is first established and then extended to larger fuels by adding relevant reaction subsets [134] [133]. For instance, a model for NH₃/DMM combustion can be built by combining a well-validated NH₃ sub-model with a dedicated DMM sub-mechanism, while carefully incorporating cross-reactions between the two fuel chemistries [129].
Sensitivity and flux analysis are indispensable tools for model interpretation. As shown in the NH₃/DMM study, reaction pathway analysis can reveal that H-abstraction reactions from the blended fuel (DMM) by radicals from the base fuel (NH₂) significantly promote ignition [129]. Furthermore, the inclusion of theoretically-predicted disproportionation reactions (radical-radical termination reactions) can be critical for accurate model prediction, as demonstrated in the same study.
A modern advancement is the move towards theory-informed kinetics, where high-level theoretical calculations are used to predict rate constants and thermochemistry, reducing reliance on empirical fitting. The Theoretically Informed Kinetics (ThInK) mechanism is a prominent example, a C0-C3 model built largely on theoretical predictions, which demonstrates improved predictive power over wide temperature and pressure ranges [134].
Computational Fluid Dynamics (CFD) Integration
Validated chemical kinetics are often integrated into CFD codes for simulating practical combustion devices. For example, the Flame Area Model (FAM) has been used to model natural gas combustion in spark-ignition engines [135]. This model solves a transport equation for a regress variable, representing the unburned gas fraction, and uses a flame wrinkling factor to account for turbulent combustion. The accuracy of such CFD simulations is fundamentally dependent on the underlying chemical kinetic model for calculating properties like laminar flame speed and ignition delay, which directly influence the predicted combustion phasing and pollutant formation [135].
The rigorous validation of chemical kinetic models using ignition delay times and species concentration profiles is a critical, non-negotiable step in developing reliable predictive tools for combustion science and reaction chemistry. This guide has detailed the experimental protocols, data standardization challenges, and advanced modeling frameworks required for this task. The iterative process of model validation and refinement, fueled by high-quality experimental data across a wide range of conditions, progressively reduces mechanism uncertainty. As the field advances, the integration of theory-informed kinetics and the application of these validated models in multi-dimensional CFD simulations will continue to enhance our fundamental understanding and engineering control over complex chemical processes, from advanced engines to novel chemical synthesis.
Computational Methods for Comparing Transition State Energies and Reaction Trajectories
Within the broader thesis on elucidating reaction mechanisms in organic chemistry research, the computational determination of transition state energies and reaction trajectories occupies a central role. For researchers and drug development professionals, these parameters are not merely theoretical constructs; they provide quantitative predictions of reaction rates, regioselectivity, and product distributions that are critical for designing efficient synthetic routes and understanding metabolic pathways [136] [137]. Transition state theory (TST) forms the foundational framework for these studies, positing that the rate constant (k) for an elementary reaction is governed by the standard Gibbs energy of activation (ΔG‡) through the Eyring equation [136] [137]. The accuracy of such predictions hinges entirely on the computational methods employed to locate transition states—first-order saddle points on the potential energy surface (PES)—and to map the minimum energy path (MEP) connecting reactants to products [138] [139]. This guide details the current computational methodologies for these tasks, providing a technical resource for advanced research.
Theoretical Foundations
Transition State Theory and the Reaction Coordinate
Transition State Theory explains reaction rates by assuming a quasi-equilibrium between reactants and an activated transition state complex [136]. The key equations are:
The Eyring Equation: k = (k₊BT/h) exp(-ΔG‡/RT) where k is the rate constant, k₊B is Boltzmann's constant, T is temperature, h is Planck's constant, ΔG‡ is the activation free energy, and R is the gas constant [136] [137].
Free Energy Components: ΔG‡ = ΔH‡ - TΔS‡ where ΔH‡ is the enthalpy of activation and ΔS‡ is the entropy of activation [136].
The reaction coordinate is a collective variable that describes the progression of structural changes along the MEP. The transition state (TS) is defined as the highest energy point on the MEP, characterized as a first-order saddle point on the PES—a minimum in all dimensions except one, along which it is a maximum [138] [139]. This structure has a Hessian matrix (the matrix of energy second derivatives) with exactly one negative eigenvalue [138].
Potential Energy Surfaces and Reaction Dynamics
The potential energy surface is a central concept in computational chemistry. A reaction mechanism is a stepwise description of the conversion of reactants to products, which may involve intermediates. The MEP is the lowest-energy pathway on this multidimensional surface connecting reactants and products [139]. For complex systems, particularly in condensed phases or enzymes, the free energy profile (A(ζ)) along a chosen order parameter (ζ) becomes crucial and is defined as: A(ζ) = -k₊BT ln(P(ζ)) + C where P(ζ) is the probability distribution of ζ, and C is a constant [140].
Computational Methodologies
Quantum Chemical Methods for Energy Calculations
The accuracy of transition state and trajectory calculations is fundamentally tied to the quantum chemical method used to compute energies on the PES.
Table 1: Comparison of Quantum Chemical Methods for TS Studies
| Method | Theoretical Basis | Accuracy | Computational Cost | Ideal Use Cases |
|---|---|---|---|---|
| AIQM2 (AI-enhanced QM) | Hybrid physical model (DFT) combined with neural network correction | Approaches coupled-cluster (CCSD(T)) accuracy for organic molecules | Orders of magnitude faster than DFT for large systems | Large-scale organic reaction simulations, reaction dynamics overnight [141] |
| Density Functional Theory (DFT) | Electron density functional | Good accuracy for many systems, depends on functional | Moderate to high, scales with system size | Workhorse for most organic and organometallic TS optimizations [141] |
| Coupled Cluster (e.g., CCSD(T)) | Wavefunction theory, gold standard | Highest accuracy for single-reference systems | Prohibitively high for large systems | Benchmark calculations for small model systems [141] |
AIQM2 represents a recent breakthrough, enabling "fast and accurate large-scale organic reaction simulations for practically relevant system sizes and time scales beyond what is possible with DFT" while maintaining robustness and transferability, avoiding catastrophic failures sometimes seen with pure machine learning potentials [141].
Locating Transition States: Algorithms and Protocols
Finding the precise geometry of a transition state is often the most challenging step. Several robust algorithms exist.
Synchronous Transit Methods
These methods use interpolated paths between reactant and product geometries as a surrogate for the MEP.
- Linear Synchronous Transit (LST): Naïvely interpolates coordinates between reactants and products and identifies the highest energy point. This often produces a poor guess with unphysical geometries and multiple imaginary frequencies [139].
- Quadratic Synchronous Transit (QST) & QST3: Uses quadratic interpolation for a more flexible constraining curve. The highest point on this curve is optimized in directions normal to the path. QST3, which requires input of reactant, product, and a guess TS geometry, is particularly robust. Even with a poor initial guess, QST3 can often recover and locate the true TS through an iterative process of redefining the constraining curve and re-optimizing [139].
Experimental Protocol for QST3 Calculation:
- Geometry Optimization: Fully optimize the structures of the reactant and product complexes.
- TS Guess Generation: Generate an initial guess for the transition state, often by manually modifying the reacting bonds or performing a relaxed coordinate scan.
- QST3 Input: Provide the optimized reactant, product, and guess TS structures as input to a QST3 algorithm (available in packages like Gaussian, ORCA).
- Iteration: The algorithm will iteratively: a. Construct a quadratic path through the three points. b. Locate the maximum along this path. c. Optimize the geometry at this maximum normal to the path. d. Use the resulting geometry as a new guess in the next cycle until convergence.
- Verification: Confirm the optimized TS has a single imaginary frequency, and that the vibrational mode corresponds to the intended reaction coordinate.
Nudged Elastic Band (NEB) and Climbing-Image NEB
Elastic band methods discretize the reaction path into a chain of structures ("images") and optimize them collectively.
- Nudged Elastic Band (NEB): A series of images are connected by spring forces along the path tangent. During optimization, only the component of the true force from the PES that is normal to the path is applied to each image, while the spring forces maintain image spacing. This "nudging" prevents the images from sliding down to the reactants or products and allows the band to find the MEP [139].
- Climbing-Image NEB (CI-NEB): An enhancement to NEB where the highest energy image is not subject to the spring forces. Instead, it is allowed to "climb" upwards along the elastic band while being minimized in directions normal to it. This forces the climbing image to the saddle point, providing a high-quality TS guess without requiring an initial TS geometry [139].
Channel Following and Dimer Methods
These methods are designed to find saddle points by following low-curvature directions on the PES.
- The Dimer Method: This method avoids Hessian calculation by using two closely spaced geometries (a "dimer") to estimate the local curvature. The dimer is rotated around its midpoint to find the direction of lowest curvature (the most negative mode). A step is then taken uphill along this direction. The process repeats until the dimer starts moving downhill, indicating a saddle point has been passed [139].
- Naïve Channel Following: This method starts from a minimum, computes the Hessian, and follows the lowest-frequency vibrational mode uphill. The Hessian is updated periodically, and the mode closest to the previous direction is followed. This is effective for small molecules but can be misled by irrelevant low-frequency modes in large systems and is computationally expensive due to repeated Hessian calculations [139].
Mapping Reaction Trajectories and Free Energies
For complex systems, especially in solution or enzymes, static transition state optimization is insufficient. Dynamic and statistical methods are required.
- Transition Path Sampling (TPS): TPS is a powerful enhanced sampling technique used to study rare barrier-crossing events in complex systems like enzymes. It uses a Monte Carlo algorithm to harvest an ensemble of true dynamical reactive trajectories without applying external bias forces. It is particularly valuable when the reaction coordinate is not known a priori. The ensemble of paths can be used to compute rate constants and elucidate mechanisms [140].
Protocol for Free Energy Calculation from TPS: A method developed by Radhakrishnan and Schlick combines TPS with a window-based sampling technique to calculate free energies [140].
- Harvest an initial ensemble of reactive trajectories using standard TPS.
- Choose an order parameter ζ that distinguishes reactants from products.
- Divide ζ into windows.
- Within each window, sample trajectories (including non-reactive ones rejected by standard TPS) using a guiding reactive trajectory.
- Combine data from all windows to construct the probability distribution P(ζ) and, via Equation 6, the free energy profile A(ζ). This approach is accurate and avoids the need for predefined collective variables as in umbrella sampling [140].
Reactive Molecular Dynamics and Analysis: Force fields like ReaxFF allow for bond formation and breaking in molecular dynamics (MD) simulations. Tools like ChemTraYzer2 can then analyze the resulting trajectories to automatically identify and count reaction events, compute reaction rate constants, and map out complex reaction networks from a single simulation, as demonstrated in studies of hydrogen/oxygen combustion [142].
Visualization and Analysis
Interpreting the vast data from computational studies requires robust visualization and analysis tools.
- Reaction Path Visualization: Tools like the open-source reaction path visualizer can generate graphical representations of complex reaction networks based on reaction fluxes from microkinetic models, helping identify dominant pathways [143].
- Trajectory Analysis: Software such as TRAVIS (Trajectory Analyzer and Visualizer) computes a wide range of properties from MD trajectories, including radial distribution functions (RDFs), spatial distribution functions (SDFs), coordination numbers, and mean-square displacements (MSDs) [144].
The following workflow diagram outlines the decision process for selecting and applying the computational methods described in this guide:
The Scientist's Toolkit
A successful computational study requires a suite of software tools and theoretical models.
Table 2: Essential Research Reagent Solutions
| Tool Category | Representative Examples | Primary Function |
|---|---|---|
| Quantum Chemistry Packages | AIQM2, Gaussian, ORCA, CP2k | Perform electronic structure calculations, geometry and TS optimizations, and frequency analyses [141] [144]. |
| Force Fields for Reactive MD | ReaxFF | Enable classical MD simulations where chemical bonds can form and break during the trajectory [142]. |
| Path Sampling & Analysis | In-house TPS codes, ChemTraYzer2 | Automate the sampling of reactive trajectories and analyze MD outputs to identify reaction events and calculate rates [142] [140]. |
| Trajectory Analyzers | TRAVIS | Compute diverse properties (RDF, SDF, MSD, vibrational spectra) from MD/MC trajectories [144]. |
| Reaction Path Visualizers | Open-source RPA visualizer | Generate graphical representations of complex reaction networks and dominant pathways [143]. |
| Transition State Theory | Eyring equation, Hammond Postulate | Provide the fundamental framework for connecting TS structure and energy to reaction kinetics and selectivity [136] [137]. |
The imperative to mitigate greenhouse gas emissions has propelled the search for sustainable, zero-carbon fuels. Within the shipping industry, this is driven by the International Maritime Organization's (IMO) 2023 strategy, which targets a peak in GHG emissions as soon as possible and net-zero emissions around 2050 [145]. Ammonia (NH₃) has emerged as a particularly promising candidate. As a zero-carbon fuel, it produces no carbon dioxide (CO₂) during combustion and offers significant advantages in storage and transportation over hydrogen [145].
However, the direct use of pure ammonia in compression-ignition engines is hindered by its high auto-ignition temperature and slow laminar burning velocity (LBV), leading to challenges in achieving stable and efficient combustion [145] [146]. A practical solution for marine engines is the dual-fuel approach, where a small amount of diesel (represented by the surrogate fuel n-heptane, NC₇H₁₆) is injected to ignite a primary charge of ammonia [145] [147]. The development and optimization of such engines rely heavily on accurate computational simulations, for which a robust understanding of the underlying reaction mechanisms is paramount [145]. This case study provides a comparative analysis of existing ammonia-n-heptane reaction mechanisms, situating this investigation within the broader context of organic chemistry research where elucidating reaction pathways is fundamental to predicting and controlling chemical outcomes.
The combustion mechanism of an ammonia-n-heptane mixture is not a simple superposition of the two individual fuel mechanisms. The interaction between carbon and nitrogen species introduces cross-reactions that critically influence the overall combustion process, such as the reaction between the fuel and the NH₂ radical, which plays a key role in determining the mixture's ignition delay time [145]. Several research groups have developed detailed and skeletal mechanisms to describe this complex kinetic network.
Table 1: Summary of Ammonia-n-Heptane Reaction Mechanisms
| Mechanism Developer(s) | Number of Species | Number of Reactions | Key Features and Development Approach |
|---|---|---|---|
| Dong et al. [145] | 2854 | 11,790 | Detailed mechanism; integrated NUIGMech1.2 with Glarborg et al.'s NH₃ sub-mechanism; incorporated C3-C7/N reactions. |
| Wang et al. [145] | 74 | 495 | Reduced mechanism; modified Dong's framework; incorporated Chang et al.'s n-heptane sub-mechanism. |
| Thorsen et al. [145] | 1367 | 6314 | Detailed mechanism; augmented Zhang et al.'s n-heptane model with Glarborg et al.'s NH₃ mechanism; added C3-C7/N reactions. |
| Fang et al. [145] | 2860 | 11,892 | Detailed mechanism; modified Dong et al.'s model; incorporated n-heptane pyrolysis intermediates' reactions and HNNO reactions. |
Comparative Performance Analysis of Different Mechanisms
Predictive Performance for Ignition Delay Time (IDT)
Ignition delay time is a critical parameter for compression-ignition engine design. Experimental data for IDT have been measured under various conditions, such as pressures of 2-25 bar, temperatures of 635-1560 K, and a wide range of ammonia blending ratios (e.g., 30% to 95% by molar content) [145] [147]. Simulations are typically performed using a zero-dimensional homogeneous closed reactor model in software like CHEMKIN-Pro, assuming a constant-volume adiabatic process. The IDT is often defined as the time corresponding to the maximum hydroxyl (OH) radical concentration [145].
The comparative analysis reveals that the four primary mechanisms show minimal discrepancies in predicting IDT across a wide range of conditions. However, significant differences emerge under high ammonia blending ratios and at elevated temperatures [145]. Studies on mixtures with low n-heptane content (2-10%) have shown that the reaction of NH₂ abstracting H atoms from n-heptane is a primary consumption pathway for n-heptane and a key ignition-promoting reaction [147]. Furthermore, the reaction NH₃ + O₂ = NH₂ + HO₂ has been identified as influential in the extent of the negative temperature coefficient (NTC) behavior observed in the mixture [147].
Predictive Performance for Oxidation and Species Concentrations
Analyzing the oxidation process and accurately predicting intermediate and product species concentrations are essential for understanding reaction pathways and validating mechanisms. Experiments often use tools like a perfectly stirred reactor (PSR) to obtain steady-state concentration data for species such as n-heptane, ammonia, oxygen, hydrogen, water, and formaldehyde at atmospheric pressure and temperatures ranging from 500-1200 K [145].
The comparative study indicates that, overall, the mechanisms can reasonably predict the concentrations of reactants and major combustion products (e.g., CO₂ and H₂O). However, significant discrepancies exist among the mechanisms in predicting the concentrations of intermediate species [145]. This highlights the need for further refinement in the subsets of the mechanisms that describe the formation and consumption of these intermediates, which is a common challenge in the validation of complex organic reaction networks.
Predictive Performance for Laminar Burning Velocity (LBV)
Laminar burning velocity is a fundamental property that characterizes flame propagation and stability. Experimental data for ammonia-n-heptane mixtures have been obtained using methods like the heat flux method and the constant volume combustion bomb (CVCB) method at pressures of 1 atm, temperatures around 338-483 K, and equivalence ratios of 0.7-1.3 [145] [146]. The CVCB method, often coupled with high-speed schlieren imaging, is valued for its ability to record the entire flame propagation process and operate at higher pressures closer to engine conditions [146].
For laminar burning velocity, the modeled values from the Dong, Thorsen, and Fang mechanisms are consistent with experimental results under both fuel-lean and fuel-rich conditions. In contrast, the Wang mechanism exhibits significant deviations from the other three in this regard [145]. Research on ammonia blended with various high-reactive fuels has confirmed that additives like n-heptane effectively enhance the otherwise slow laminar flame speed of pure ammonia [146].
Diagram 1: Mechanism validation workflow.
Reaction Pathway and Sensitivity Analysis
In-depth analysis of reaction pathways and sensitivity is crucial for understanding the fundamental kinetic differences between mechanisms. Such analysis aligns with core practices in organic chemistry, where mechanistic studies aim to deconstruct complex reactions into elementary steps to explain product formation and kinetics.
Pathway analysis reveals that the Wang mechanism exhibits significant deviations from the other three in predicting the detailed reaction pathways of ammonia and n-heptane [145]. Despite this, when viewed from the perspective of reaction class, the mechanisms are somewhat similar in the key reactions governing the consumption of the two fuels [145].
Sensitivity analysis helps identify the specific reactions that have the greatest impact on combustion properties like ignition. For ammonia-n-heptane mixtures with low n-heptane content, the reaction of ammonia consuming OH radicals (NH₃ + OH = NH₂ + H₂O) is identified as the most inhibiting reaction for ignition [147]. This underscores the critical role of cross-reactions between the nitrogen-based and carbon-based chemical families.
Diagram 2: Key ammonia consumption pathways.
Application in Engine Simulation and Mechanism Selection
From a practical application standpoint, the ultimate test of a chemical mechanism is its performance in simulating real-world combustion devices, such as internal combustion engines. When the four mechanisms are incorporated into a multi-zone engine model to predict in-cylinder pressure and temperature profiles, the results show minimal variations across the different mechanisms [145].
This finding has significant practical implications for engineers. It suggests that for the specific purpose of engine performance simulation, where computational cost is a major concern, a reduced mechanism can be sufficiently accurate. The study concludes that the Fang mechanism is the most accurate choice for fundamentally understanding the ignition, oxidation, and flame characteristics of ammonia-n-heptane mixtures. However, to reduce computational cost in full-engine simulations, the Wang mechanism (with only 74 species and 495 reactions) is a viable and efficient alternative [145].
The Scientist's Toolkit: Research Reagents and Experimental Essentials
Table 2: Key Research Reagents and Experimental Materials
| Item / Solution | Function / Role in Investigation |
|---|---|
| n-Heptane (NC₇H₁₆) | A single-component surrogate fuel for diesel due to its similar cetane number; simplifies the complex chemical system for foundational studies [145]. |
| Anhydrous Ammonia (NH₃) | The primary zero-carbon fuel under investigation; its high purity is essential to avoid confounding effects from water or other impurities [146]. |
| Constant Volume Combustion Bomb (CVCB) | An experimental apparatus used to measure laminar burning velocity under conditions of elevated pressure, mimicking those in internal combustion engines [146]. |
| Rapid Compression Machine (RCM) | A device used to experimentally measure ignition delay times at elevated temperatures and pressures relevant to engine compression ignition [147]. |
| Perfectly Stirred Reactor (PSR) Model | A computational tool within chemical kinetics software (e.g., CHEMKIN-Pro) used to simulate fuel oxidation and obtain steady-state species concentration data [145]. |
| High-Speed Schlieren Imaging System | An optical diagnostic technique used with CVCBs to visually track flame kernel growth and instability during spherical flame propagation [146]. |
This comparative analysis of ammonia-n-heptane reaction mechanisms underscores the nuanced balance between chemical fidelity and computational tractability in reaction kinetics. While detailed mechanisms like those from Fang and Dong et al. offer superior accuracy in predicting fundamental combustion properties like ignition delay and intermediate species, reduced mechanisms such as Wang's provide a computationally efficient alternative suitable for system-level engineering simulations. The findings highlight that the C-N cross-reactions are central to accurately modeling the combustion behavior. This case study exemplifies the broader paradigm in organic chemistry and fuel research, where the development and validation of multi-step reaction mechanisms are indispensable for transitioning from fundamental chemical insight to practical technological application. The continued refinement of these mechanisms, guided by targeted experimental data, will be critical for the successful development of ammonia-powered, low-carbon marine engines.
Establishing the Applicability Domain of a Proposed Reaction Mechanism
In organic chemistry research, proposing a reaction mechanism is a fundamental step towards understanding chemical transformations. However, the true utility and reliability of a mechanistic proposal are determined by rigorously establishing its applicability domain (AD)—the specific conditions and types of reactions for which the mechanism provides a valid and accurate description [148]. A well-defined AD is crucial for researchers and drug development professionals to confidently apply a mechanism in new contexts, such as predicting reaction outcomes in synthetic routes or understanding metabolic pathways in pharmaceutical development. Without a clear AD, extrapolating a mechanism beyond its validated boundaries risks incorrect predictions and failed experiments. This guide provides a comprehensive framework for establishing the AD of a proposed reaction mechanism, integrating both theoretical concepts and practical experimental and computational protocols.
Defining the Applicability Domain for Reaction Mechanisms
The applicability domain of a reaction mechanism defines the chemical space where its predictions are considered reliable. This space is bounded by the structural features of the reactants, the reaction conditions, and the types of chemical transformations for which the mechanism has been validated [148].
For reaction mechanisms, the AD must encompass several critical dimensions, which can be visualized as a multi-faceted validation space. The subsequent diagram illustrates the core workflow for establishing this domain, highlighting the interplay between computational and experimental validation.
Key Dimensions of an Applicability Domain for Reaction Mechanisms:
- Structural Domain: The range of reactant and catalyst structures (including functional groups, steric properties, and electronic characteristics) for which the mechanism holds true.
- Conditional Domain: The ranges of temperature, pressure, solvent, pH, and concentration over which the mechanism operates without change.
- Transformational Domain: The specific types of chemical transformations (e.g., nucleophilic substitution, pericyclic reaction) and their variations that follow the proposed pathway [148].
Establishing the AD involves demonstrating that the mechanism consistently explains all experimental observations within these boundaries and identifying where it begins to break down, often through systematic variation of reaction parameters and computational exploration of the potential energy surface.
Methodologies for Establishing Applicability Domain
A multi-technique approach, combining computational and experimental methods, is essential for rigorously defining the AD of a reaction mechanism. The following table summarizes the key methodologies, their implementation, and the specific AD dimension they probe.
Table 1: Key Methodologies for Establishing the Applicability Domain of a Reaction Mechanism
| Methodology | Implementation | Primary AD Dimension Probed |
|---|---|---|
| Computational Potential Energy Surface (PES) Exploration | Use AIQM2 [149] or DFT to locate transition states and intermediates; calculate reaction barriers and energies for varied substrates. | Transformational Domain, Structural Domain |
| Kinetic Analysis | Determine rate law and order for each reactant; measure activation parameters (Eₐ, ΔH‡, ΔS‡) via variable temperature studies [93]. | Transformational Domain, Conditional Domain |
| Reaction Intermediate Detection | Use spectroscopic methods (IR, NMR, EPR) or chemical trapping to detect/characterize proposed intermediates [93]. | Transformational Domain |
| Stereochemical Studies | Determine stereochemistry of products from stereodefined starting materials [93]. | Transformational Domain |
| Isotopic Labeling | Incorporate isotopes (e.g., ²H, ¹³C, ¹⁸O) at specific positions and track their fate in products [93]. | Transformational Domain |
| Applicability Domain Algorithms | Apply leverage, Z-1NN, or bounding box methods to reaction descriptor space to identify reliable prediction zones [148] [150]. | Structural Domain, Conditional Domain |
Computational PES Exploration with AI-Enhanced Methods
Advanced computational methods can map the potential energy surface (PES) to validate a mechanism's feasibility and explore its scope.
- AI-Enhanced Quantum Mechanics (e.g., AIQM2): The AIQM2 method provides a breakthrough for reaction simulations, offering accuracy approaching the gold-standard coupled-cluster level at a computational cost orders of magnitude lower than common DFT [149]. Its speed enables large-scale screening across the structural and transformational domain.
- Protocol:
- Core Mechanism Validation: Optimize the geometries of proposed reactants, products, transition states, and intermediates for a representative model system. Calculate the associated reaction energies and barrier heights.
- Structural Domain Probing: Systematically vary the substituents on the core reactants (e.g., electron-donating vs. electron-withdrawing groups, steric bulk) and re-optimize the critical points on the PES. Significant changes in the mechanism (e.g., new intermediates, different rate-determining steps) indicate a boundary of the AD.
- Uncertainty Estimation: Leverage AIQM2's built-in uncertainty estimates to identify regions of chemical space where the predictions are less reliable, signaling the edge of the AD [149].
- Protocol:
Experimental Validation Protocols
Experimental data is irreplaceable for validating computational predictions and defining the AD.
Protocol for Kinetic Analysis to Determine Reaction Order [93]:
- Initial Rate Method: Perform the reaction multiple times, varying the concentration of one reactant while keeping others in large excess. Measure the initial rate for each experiment.
- Data Analysis: Plot the logarithm of the initial rate against the logarithm of the varied reactant's concentration. The slope of the resulting line is the reaction order with respect to that reactant.
- AD Interpretation: A change in reaction order for a key reactant when moving to a different structural family of substrates indicates a fundamental change in mechanism, marking a boundary of the AD.
Protocol for Isotopic Labeling Studies [93]:
- Design: Synthesize a reactant labeled with a stable isotope (e.g., Deuterium for Hydrogen, ¹³C for ¹²C) at a specific position proposed to be involved in the mechanism (e.g., a bond cleavage site).
- Execution: Run the reaction with the labeled compound and isolate the products.
- Analysis: Use techniques like NMR or Mass Spectrometry to determine the location of the isotope in the products.
- AD Interpretation: A different isotopic distribution in products obtained from a modified reaction system (e.g., different solvent or catalyst) signals a change in the mechanism's pathway.
Formal Applicability Domain Algorithms
Formal AD algorithms from chemoinformatics can be adapted to define the boundaries for a reaction mechanism model.
- Leverage Method: This approach identifies if a new reaction is structurally dissimilar to those used to establish the mechanism.
- Algorithm: The leverage ( h ) of a test reaction is calculated as ( h = xi^T(X^TX)^{-1}xi ), where ( X ) is the descriptor matrix of the training set reactions, and ( x_i ) is the descriptor vector for the test reaction [148]. A reaction with a leverage higher than a threshold (e.g., ( h^* = 3p/n ), where ( p ) is the number of descriptors and ( n ) is the number of training reactions) is considered an X-outlier and outside the AD [148] [150].
- Distance-Based Methods (e.g., Z-1NN): This method evaluates the distance of a test reaction from its nearest neighbor in the training set.
- Algorithm: The Euclidean distance ( D ) between the test reaction and its nearest neighbor in the training set is calculated. If ( D > Z\sigma + \langle y \rangle ), where ( \sigma ) is the standard deviation of nearest-neighbor distances in the training set and ( Z ) is a parameter (often 0.5), the reaction is an X-outlier [148].
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key reagents and materials essential for conducting the experimental validation of a reaction mechanism's applicability domain.
Table 2: Key Research Reagent Solutions for Mechanistic Studies
| Reagent / Material | Function in Mechanistic Studies |
|---|---|
| Isotopically Labeled Compounds (e.g., D₂O, ¹³C-labeled substrates) | To trace atom fate and determine kinetic isotope effects (KIE) for probing transition state structure and pathway [93]. |
| Radical Traps (e.g., TEMPO, BHT) | To detect the involvement of radical intermediates; reaction inhibition suggests a radical pathway. |
| Chemical Quenching Agents | To rapidly stop a reaction at various time points for analysis, enabling intermediate detection and kinetic profiling. |
| Chiral Stationary Phases (for HPLC/GC) | To separate and analyze enantiomers for stereochemical studies, determining inversion or retention of configuration. |
| Deuterated Solvents (e.g., CDCl₃, DMSO-d₆) | As the medium for in-situ reaction monitoring and intermediate characterization via NMR spectroscopy. |
| In-situ Spectroscopy Cells (e.g., ATR-IR, ReactR) | To provide real-time data on reaction progress and intermediate formation for kinetic analysis. |
An Integrated Workflow: Putting It All Together
Establishing the AD is an iterative process. The following Graphviz diagram details the experimental protocol for kinetic analysis, a cornerstone of mechanistic validation, showing how data feeds into AD definition.
This integrated approach, combining computational power with rigorous experimental validation, ensures that a proposed reaction mechanism is not just a theoretical construct but a robust, well-defined model with clear boundaries for its application. This is fundamental for advancing predictive organic chemistry and accelerating efficient drug development.
Conclusion
A deep and practical understanding of organic reaction mechanisms is indispensable in modern drug discovery and development. This synthesis has demonstrated that moving from foundational principles to application, optimization, and rigorous validation creates a powerful framework for innovation. The ability to rationally design and troubleshoot synthetic routes, exemplified by the pivotal role of cross-coupling reactions, directly accelerates the creation of clinical candidates. Looking forward, the integration of advanced computational models, artificial intelligence, and sophisticated validation techniques will further enhance our predictive control over complex reactions. This evolving mechanistic understanding will continue to be a critical driver for developing new therapeutic modalities, ultimately enabling the discovery of more effective and sophisticated medicines to address unmet medical needs.
References
- 1. Reaction mechanisms - Latest research and news [https://www.nature.com/subjects/reaction-mechanisms]
- 2. 6.3: Bond Breaking and Bond Making [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Smith)/06%3A_Understanding_Organic_Reactions/6.03%3A_Bond_Breaking_and_Bond_Making]
- 3. Electron Pushing in Organic Chemistry [https://organicchemistrydata.org/hansreich/resources/electron_pushing/]
- 4. Rethinking the Chemical Reaction as a Graph - Depth-First [https://depth-first.com/articles/2020/02/24/rethinking-the-chemical-reaction-as-a-graph-imaginary-transition-structures-and-beyond/]
- 5. Current research, reaction mechanisms, and future ... [https://www.sciencedirect.com/science/article/abs/pii/S0360319925039254]
- 6. 2025 Inorganic Reaction Mechanisms Conference GRC [https://www.grc.org/inorganic-reaction-mechanisms-conference/2025/]
- 7. Comparing The SN1 vs Sn2 Reactions [https://www.masterorganicchemistry.com/2012/08/08/comparing-the-sn1-and-sn2-reactions/]
- 8. SN1 and SN2 reaction – Kinetics, Mechanism ... [https://chemicalnote.com/sn1-and-sn2-reaction-kinetics-mechanism-stereochemistry-and-reactivity/]
- 9. SN1 versus SN2 Reactions, Factors Affecting SN1 and ... [https://www.pharmaguideline.com/2021/11/sn1-versus-sn2-reactions-factors-affecting-nas2-and-ns2-reactions.html]
- 10. SN1 vs. SN2 Explained: Rate Laws, Reactivity, and Solvent ... [https://eureka.patsnap.com/blog/sn1-vs-sn2-reaction-mechanisms/]
- 11. Deciding SN1/SN2/E1/E2 (1) - The Substrate [https://www.masterorganicchemistry.com/2012/11/21/deciding-sn1sn2e1e2-1-the-substrate/]
- 12. SN1 and SN2 Reactions - Kinetics, Order of Reactivity ... [https://www.pharmaguideline.com/2021/11/sn1-and-sn2-reactions.html]
- 13. Mechanism of the E2 Reaction [https://www.masterorganicchemistry.com/2012/09/27/the-e2-mechanism/]
- 14. 2.9: The Mechanism of the E2 Reaction [https://chem.libretexts.org/Courses/Smith_College/CHM_223_Chemistry_III%3A_Organic_Chemistry_(2025)/02%3A_Reactions_of_Alkyl_Halides-_Nucleophilic_Substitutions_and_Eliminations/2.09%3A_The_Mechanism_of_the_E2_Reaction]
- 15. E1 vs E2: Comparing the E1 and E2 Reactions [https://www.masterorganicchemistry.com/2012/10/10/comparing-the-e1-and-e2-reactions/]
- 16. The E1 Reaction and Its Mechanism [https://www.masterorganicchemistry.com/2012/09/19/the-e1-reaction/]
- 17. 3 Factors That Stabilize Carbocations [https://www.masterorganicchemistry.com/2011/03/11/3-factors-that-stabilize-carbocations/]
- 18. Carbocation Stability [https://www.chemistrysteps.com/stability-of-carbocations/]
- 19. Intermolecular 1,2-difunctionalization of alkenes [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d5cs00670h]
- 20. Reactive Intermediates: Carbocations, Carbanions, Free ... [https://www.igi-global.com/chapter/reactive-intermediates/382126]
- 21. Identifying Intermediates in a Reaction Mechanism [https://www.bocsci.com/resources/identifying-intermediates-in-a-reaction-mechanism-methods-examples.html?srsltid=AfmBOoo55g2Btl4bf8p6WjDAzv360jbP7nEPtOeI8lXmzyD8RnqARERn]
- 22. Stability Order of Carbocation, Carbanion and Free Radicals [https://onlineorganicchemistrytutor.com/stability-order-carbocation-carbanion-free-radicals/]
- 23. Reaction Intermediates: Radical, Carbocation, Carbanion [https://www.studyorgo.com/blog/reaction-intermediates-radical-carbocation-carbanion/]
- 24. spatio-temporal visualization of reactive carbocation ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc06447c]
- 25. Reactive Intermediates in Organic Synthesis [https://www.acs.org/events/spring/digital-meeting/global-virtual-symposia/reactive-intermediates-in-organic-synthesis.html]
- 26. 2025 Organic Reactions and Processes Conference GRC [https://www.grc.org/organic-reactions-and-processes-conference/2025/]
- 27. Wagner–Meerwein rearrangement - Wikipedia [https://en.wikipedia.org/wiki/Wagner%E2%80%93Meerwein_rearrangement]
- 28. 30.1: Wagner-Meerwein Rearrangements - Chemistry LibreTexts [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Book:_Virtual_Textbook_of_OChem_(Reusch)_UNDER_CONSTRUCTION/30:_Cationic_Rearrangements/30.1:_Wagner-Meerwein_Rearrangements]
- 29. Rearrangement Reactions (1) - Hydride Shifts [https://www.masterorganicchemistry.com/2012/08/15/rearrangement-reactions-1-hydride-shifts/]
- 30. A unified approach to highly functionalized clovane-type ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04197j]
- 31. Pericyclic reaction [https://en.wikipedia.org/wiki/Pericyclic_reaction]
- 32. Pericyclic Reactions [https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/pericycl.htm]
- 33. 1.1: Introduction to Pericyclic Reactions [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Synthesis_(Shea)/01%3A_Pericyclic_Reactions/1.01%3A_Introduction_to_Pericyclic_Reactions]
- 34. Pericyclic reactions catalyzed by chorismate-utilizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3164438/]
- 35. Experimental Techniques to Study Organic Mechanisms [https://www.mckgroup.org/experimental-techniques-to-study-organic-chemistry-mechanisms]
- 36. Isotope Labeling Techniques: A Comprehensive Guide ... [https://www.metwarebio.com/isotope-labeling-guide-applications/]
- 37. Liver Microsomal Fraction induced Small Molecule Isotope ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267025011997]
- 38. Advances in Isotope Labeling for Solution Nucleic Acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12261052/]
- 39. Flow chemistry as a tool for high throughput experimentation [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00129c]
- 40. Emerging trends in the optimization of organic synthesis ... [https://www.sciencedirect.com/science/article/pii/S1860539725000088]
- 41. Generic Machine Learning Potential for Accelerating ... [https://chemrxiv.org/engage/chemrxiv/article-details/68cbca7c23be8e43d6d2503a]
- 42. What's A Transition State? [https://www.masterorganicchemistry.com/2010/11/03/whats-a-transition-state/]
- 43. Finding Transition Structures [https://server.ccl.net/cca/documents/dyoung/topics-orig/ts.html]
- 44. How to Draw Transition States - Organic Chemistry Tutor [https://www.organicchemistrytutor.com/topic/how-to-draw-transition-states/]
- 45. Constructing Transition States [https://learn.openochem.org/learn/first-semester-topics/acidsbase-and-reactions/energy-diagrams-and-reactions/constructing-transition-states]
- 46. making sense of multiple transition states in parallel and ... [https://pubs.rsc.org/en/content/articlelanding/2025/cs/d4cs00868e]
- 47. New model predicts a chemical reaction's point of no return [https://www.sciencedaily.com/releases/2025/04/250423111900.htm]
- 48. Conformational dynamics of 4-formylaminoantipyrine ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286018302746]
- 49. A Comprehensive Computational NMR Analysis of Organic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10608599/]
- 50. Transition state structure detection with machine learningś [https://www.nature.com/articles/s41524-025-01693-4]
- 51. Coupling reaction [https://en.wikipedia.org/wiki/Coupling_reaction]
- 52. Recent advances in palladium-catalyzed Suzuki-Miyaura ... [https://www.sciencedirect.com/science/article/abs/pii/S0022328X25000634]
- 53. A reactivity model for oxidative addition to palladium enables ... [https://pubs.rsc.org/en/content/articlelanding/2022/sc/d2sc00174h]
- 54. Predicting success in Cu-catalyzed C–N coupling reactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10793951/]
- 55. 17.2. Palladium catalyzed couplings | Organic Chemistry II [https://courses.lumenlearning.com/suny-potsdam-organicchemistry2/chapter/17-2-palladium-catalyzed-couplings/]
- 56. Suzuki Coupling [https://www.organic-chemistry.org/namedreactions/suzuki-coupling.shtm]
- 57. Development of a Buchwald-Hartwig Amination for an ... [https://pubmed.ncbi.nlm.nih.gov/39811117/]
- 58. Suzuki–Miyaura (hetero-)aryl cross-coupling [https://pubs.rsc.org/en/content/articlelanding/2025/cs/d4cs01108b]
- 59. Adaptive experimentation and optimization in organic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599388/]
- 60. Losartan in 2025: Pharmacology, Guidelines, and Practical ... [https://wirelesslifesciences.org/losartan-in-2025-pharmacology-guidelines-and-practical-prescribing/]
- 61. An Eco-Friendly Approach to Losartan Synthesis Using Green ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12155655/]
- 62. Cyclin-dependent kinase inhibitor abemaciclib-induced ... [https://pubmed.ncbi.nlm.nih.gov/40865691/]
- 63. Losartan - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/chemistry/losartan]
- 64. Lilly to present new clinical data for Verzenio (abemaciclib ... [https://www.prnewswire.com/news-releases/lilly-to-present-new-clinical-data-for-verzenio-abemaciclib-and-multiple-novel-pipeline-programs-at-the-2025-european-society-for-medical-oncology-esmo-annual-meeting-302581179.html]
- 65. Verzenio (Abemaciclib) a New CDK4/CDK6 Inhibitor ... [https://www.jhoponline.com/issues/special-issues/verzenio-abemaciclib-a-new-cdk4-cdk6-inhibitor-approved-for-hr-positive-her2-negative-advanced-or-metastatic-breast-cancer]
- 66. Risk factors for adverse reactions caused by abemaciclib in ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1529980/full]
- 67. An overview of PROTACs: a promising drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9763089/]
- 68. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 69. Natural Cyclopeptides as Anticancer Agents in the Last 20 Years [https://pmc.ncbi.nlm.nih.gov/articles/PMC8068844/]
- 70. Various mechanisms in cyclopeptide production from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3180235/]
- 71. Cyclopeptide - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/cyclopeptide]
- 72. DNA-Encoded Chemical Libraries: A Comprehensive Review ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8369695/]
- 73. DNA-encoded chemical library [https://en.wikipedia.org/wiki/DNA-encoded_chemical_library]
- 74. Driving Drug Design Using DNA Encoded Libraries [https://www.amgen.com/stories/2025/04/driving-drug-design-using-dna-encoded-libraries]
- 75. Recent advances in DNA-encoded libraries: From covalent ... [https://www.sciencedirect.com/science/article/pii/S0959440X25001812]
- 76. Structure–activity relationship [https://en.wikipedia.org/wiki/Structure%E2%80%93activity_relationship]
- 77. Structure-Activity Relationship - an overview [https://www.sciencedirect.com/topics/chemistry/structure-activity-relationship]
- 78. On Exploring Structure Activity Relationships - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4852705/]
- 79. Key Concepts of Structure-Activity Relationships to Know ... [https://fiveable.me/lists/key-concepts-of-structure-activity-relationships]
- 80. Structure Activity Relationship (SAR) Analysis [https://www.creative-proteomics.com/services/structure-activity-relationship-sar-analysis.htm]
- 81. Comparison of Quantitative and Qualitative (Q)SAR Models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6192375/]
- 82. Comparative Study of (Q)SAR Models for Predicting the ... [https://www.sciencedirect.com/science/article/pii/S3050620425000491]
- 83. RMG - Reaction Mechanism Generator [https://rmg.mit.edu/]
- 84. Organic Syntheses Procedure [http://orgsyn.org/demo.aspx?prep=cv6p0218]
- 85. Organic Syntheses Procedure [http://orgsyn.org/demo.aspx?prep=v97p0207]
- 86. Organic Syntheses Procedure [http://orgsyn.org/demo.aspx?prep=v94p0016]
- 87. ACE Reaction Network — Tutorials 2025.1 documentation [https://www.scm.com/doc/Tutorials/WorkflowsAndAutomation/ReactionNetwork.html]
- 88. Ansys Chemkin | Chemical Kinetics Simulation Software [https://www.ansys.com/products/fluids/ansys-chemkin]
- 89. Kintecus - Enzyme & Combustion Chemical Kinetics and ... [https://www.kintecus.com/]
- 90. Top 6 Visualizations for Quantitative Data Analysis Methods [https://chartexpo.com/blog/quantitative-data-analysis-methods]
- 91. Reaction-conditioned generative model for catalyst design ... [https://www.nature.com/articles/s42004-025-01732-7]
- 92. Data-driven discovery of active phosphine ligand space for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11339946/]
- 93. Reaction mechanism [https://en.wikipedia.org/wiki/Reaction_mechanism]
- 94. Organic Syntheses Procedure [https://orgsyn.org/demo.aspx?prep=CV6P0919]
- 95. Integrative catalytic pairs driving complex chemical reactions [https://www.nature.com/articles/s41570-025-00771-x]
- 96. Covalent organic and metal organic frameworks based single ... [https://pubs.rsc.org/en/content/articlehtml/2025/ee/d5ee02702k]
- 97. Stereoselective and Stereospecific Reactions [https://www.masterorganicchemistry.com/2010/07/02/stereoselective-stereospecific/]
- 98. Asymmetric induction [https://en.wikipedia.org/wiki/Asymmetric_induction]
- 99. Alkene Addition Reactions: "Regioselectivity" and " ... [https://www.masterorganicchemistry.com/2013/01/22/alkene-addition-regioselectivity-syn-anti/]
- 100. Significance and challenges of stereoselectivity assessing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5762452/]
- 101. Mechanism and origin of stereoselectivity and regioselectivity ... [https://pubs.rsc.org/en/content/articlehtml/2025/cy/d5cy00519a]
- 102. A density functional theory study [https://www.sciencedirect.com/science/article/abs/pii/S2210271X25000222]
- 103. Computational tools for the prediction of site - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc00541h]
- 104. Full Regio- and Stereoselective Protocol for the Synthesis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10142438/]
- 105. Theoretical Study on the Origin of Abnormal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9962681/]
- 106. Are activation barriers of 50–70 kcal mol−1 accessible for ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc08243e]
- 107. New modified Arrhenius equation to describe the ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725041348]
- 108. Kinetics of the reaction of CF 3 CHO with OH between 204 ... [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp02871j]
- 109. Reaction behavior and kinetic modeling for high- ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250925018238]
- 110. Large language model guided automated reaction ... [https://www.nature.com/articles/s42004-025-01630-y]
- 111. Modeling solvent effects in chemical reactions [https://communities.springernature.com/posts/modeling-solvent-effects-in-chemcial-reactions]
- 112. Advanced green synthesis: Solvent-free and catalyst ... [https://www.sciencedirect.com/science/article/pii/S2666554924001285]
- 113. Experimental and computational analysis of lipophilicity ... [https://www.sciencedirect.com/science/article/abs/pii/S1570023225000339]
- 114. Fsp3: A new parameter for drug-likeness [https://pubmed.ncbi.nlm.nih.gov/32712310/]
- 115. H functionalization via molecular electrocatalysis [https://pubs.rsc.org/en/content/articlelanding/2025/qo/d5qo01036e]
- 116. Recent synthetic methodology advancements towards all ... [https://www.sciencedirect.com/science/article/abs/pii/S0040402025002674]
- 117. From Patterns to Pills: How Informatics Is Shaping Medicinal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114992/]
- 118. How To: Troubleshoot a Reaction [https://www.chem.rochester.edu/notvoodoo/pages/how_to.php?page=experiment]
- 119. Planning Organic Synthesis With "Reaction Maps" [https://www.masterorganicchemistry.com/2013/12/17/introduction-to-synthesis/]
- 120. 4.7: Reaction Work-Ups [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_Lab_Techniques_(Nichols)/04%3A_Extraction/4.07%3A_Reaction_Work-Ups]
- 121. Process Validation in Six Sigma: A Complete Guide [https://www.6sigma.us/process-improvement/process-validation/]
- 122. Food for Thought ... Mechanistic Validation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4086802/]
- 123. ORGANIC 2025 | August 18-19, 2025 | Valencia | Spain [https://organic.chemistryconferences.org/events-list/organic-reaction-mechanisms-and-dynamics]
- 124. A large-scale reaction dataset of mechanistic of pathways ... organic [https://www.nature.com/articles/s41597-024-03709-y?error=cookies_not_supported&code=c2bbc7d1-b496-43cc-baf6-96f305e42201]
- 125. Data-driven recommendation of agents, temperature, and ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d5sc04957a]
- 126. Enhancing students’ agency and coherence in organic ... chemistry [https://pubs.rsc.org/en/content/articlehtml/2025/rp/d5rp00268k]
- 127. Research on the reaction mechanism and molecular ... [https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp01024a]
- 128. oMeBench: Towards Robust Benchmarking of LLMs in Organic ... [https://arxiv.org/html/2510.07731v2]
- 129. Shock tube and modeling study on the ignition delay times ... [https://www.sciencedirect.com/science/article/abs/pii/S0010218023003437]
- 130. Standardizing the Ignition Delay Time Measurements of ... [https://www.mdpi.com/1996-1073/18/1/165]
- 131. An experimental and kinetic modeling study of the ignition ... [https://www.sciencedirect.com/science/article/abs/pii/S0010218024005935]
- 132. Modeling Temperature and Species Concentration Profiles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9202279/]
- 133. A comprehensive review of detailed kinetics models for ... [https://link.springer.com/article/10.1007/s43153-025-00606-8]
- 134. Establishing a modern C0-C3 mechanism for combustion ... [https://www.sciencedirect.com/science/article/abs/pii/S0010218025005383]
- 135. Development and Validation of a CFD Combustion Model ... [https://www.mdpi.com/1996-1073/16/2/971]
- 136. Transition state theory [https://en.wikipedia.org/wiki/Transition_state_theory]
- 137. reaction rates, transition states, and mechanisms [https://research.cm.utexas.edu/nbauld/unit2.htm]
- 138. Transition-state theory — Computational Chemistry from ... [https://kthpanor.github.io/echem/docs/mol_struct/tst.html]
- 139. Guessing Transition States - Rowan Scientific [https://rowansci.com/blog/guessing-transition-states]
- 140. Path Sampling Based Calculations of Free Transition for... Energies [https://pmc.ncbi.nlm.nih.gov/articles/PMC9444332/]
- 141. AIQM2: organic reaction simulations beyond DFT - Chemical Science... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d5sc02802g]
- 142. ChemTraYzer2: Reactive MD Analysis — Tutorials ... - SCM [https://www.scm.com/doc/Tutorials/MolecularDynamicsAndMonteCarlo/ChemTrayzer2.html]
- 143. An open-source framework for reaction path visualization ... [https://www.sciencedirect.com/science/article/pii/S2352711019302432]
- 144. TRAVIS – Trajectory Analyzer and Visualizer [https://brehm-research.de/travis.php]
- 145. From Ignition, Oxidation, and Laminar Flame Propagation ... [https://www.mdpi.com/2571-6255/8/9/357]
- 146. Study on the Combustion Characteristics and Reaction ... [https://www.sciencedirect.com/science/article/abs/pii/S1743967125002983]
- 147. Auto-Ignition Characteristics and Chemical Reaction ... [https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4617752]
- 148. Comprehensive Analysis of Applicability Domains of QSPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7432167/]
- 149. AIQM2: organic reaction simulations beyond DFT [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc02802g]
- 150. On a simple approach for determining applicability domain ... [https://www.sciencedirect.com/science/article/abs/pii/S0169743915000969]