Stereochemistry and Isomerism in Drug Development: From Molecular Principles to Clinical Applications
This article provides a comprehensive examination of stereochemistry and isomerism for researchers and professionals in drug development.
Stereochemistry and Isomerism in Drug Development: From Molecular Principles to Clinical Applications
Abstract
This article provides a comprehensive examination of stereochemistry and isomerism for researchers and professionals in drug development. It covers the foundational principles of stereoisomers, enantiomers, and diastereomers, explores advanced methodological approaches for stereochemical analysis and resolution, addresses common challenges in bioanalytical method development, and discusses rigorous validation techniques for absolute configuration determination. The content highlights the critical impact of molecular chirality on drug safety, efficacy, and regulatory compliance, with practical insights from pharmaceutical case studies including thalidomide, beta-lactams, and chiral switches.
Fundamental Principles of Molecular Symmetry and Isomerism
Stereochemistry, the study of the three-dimensional arrangement of atoms in molecules, is a fundamental discipline with profound implications across chemistry, biology, and medicine. This whitepaper delineates the core principles of stereochemistry, exploring how molecular geometry dictates properties and functions in chemical and biological systems. Within the broader context of isomerism research, we examine how stereoisomers—identical in atomic connectivity but divergent in spatial orientation—exhibit dramatically different biological activities, physical properties, and reactivities. For researchers and drug development professionals, mastering stereochemistry is not merely academic but crucial for designing effective pharmaceuticals, understanding biochemical pathways, and developing advanced materials. This guide provides a technical foundation of stereochemical concepts, supported by quantitative data, experimental protocols, and analytical methodologies essential for contemporary research and development.
Stereochemistry is the branch of chemistry concerned with the three-dimensional (3D) arrangement of atoms and molecules and how this spatial configuration influences their properties and reactions [1]. It is often described as the "chemistry of space," stemming from the Greek word "stereos," meaning solid or three-dimensional [2]. In the broader research landscape of molecular isomerism, stereochemistry specifically addresses stereoisomers—molecules that share the same molecular formula and atomic connectivity (bonding sequence) but differ in the orientation of their atoms in space [2].
This spatial arrangement is critical because it directly governs how a molecule interacts with biological targets, its chemical reactivity, and its physical properties. In essence, the 3D structure of a molecule is as fundamental to its identity and function as its 2D connectivity. For drug development professionals, this is paramount; the pharmacological activity of a compound is often exclusively dependent on its correct stereochemical configuration [3]. A molecule with the "wrong" handedness may be inactive, less potent, or even cause toxic side effects, as tragically demonstrated by the drug thalidomide [4].
The study of stereochemistry intersects with all domains of chemistry—organic, inorganic, and physical—but is particularly critical in organic chemistry and biochemistry, where chiral molecules are ubiquitous. Understanding stereochemistry allows scientists to rationalize reaction outcomes, design asymmetric syntheses, and predict the behavior of molecules in complex environments, from catalytic systems to living organisms.
Fundamental Concepts and Classification of Stereoisomers
At the heart of stereochemistry lies the concept of chirality. A molecule is considered chiral if it cannot be superimposed on its mirror image, much like a left and right hand [3] [2]. This property most commonly arises from a chiral center, typically a carbon atom with four different substituents [3]. The two non-superimposable mirror images of a chiral molecule are called enantiomers [1].
Table 1: Fundamental Types of Stereoisomers
| Type | Definition | Key Characteristics | Example |
|---|---|---|---|
| Enantiomers | Mirror images that are not superimposable [2]. | Have identical physical properties except for the direction they rotate plane-polarized light; often exhibit different biological activities [3]. | D- and L-glucose [5]. |
| Diastereomers | Stereoisomers that are not mirror images [2]. | Have different physical and chemical properties [2]. | cis- and trans- isomers; different forms of tartaric acid [2]. |
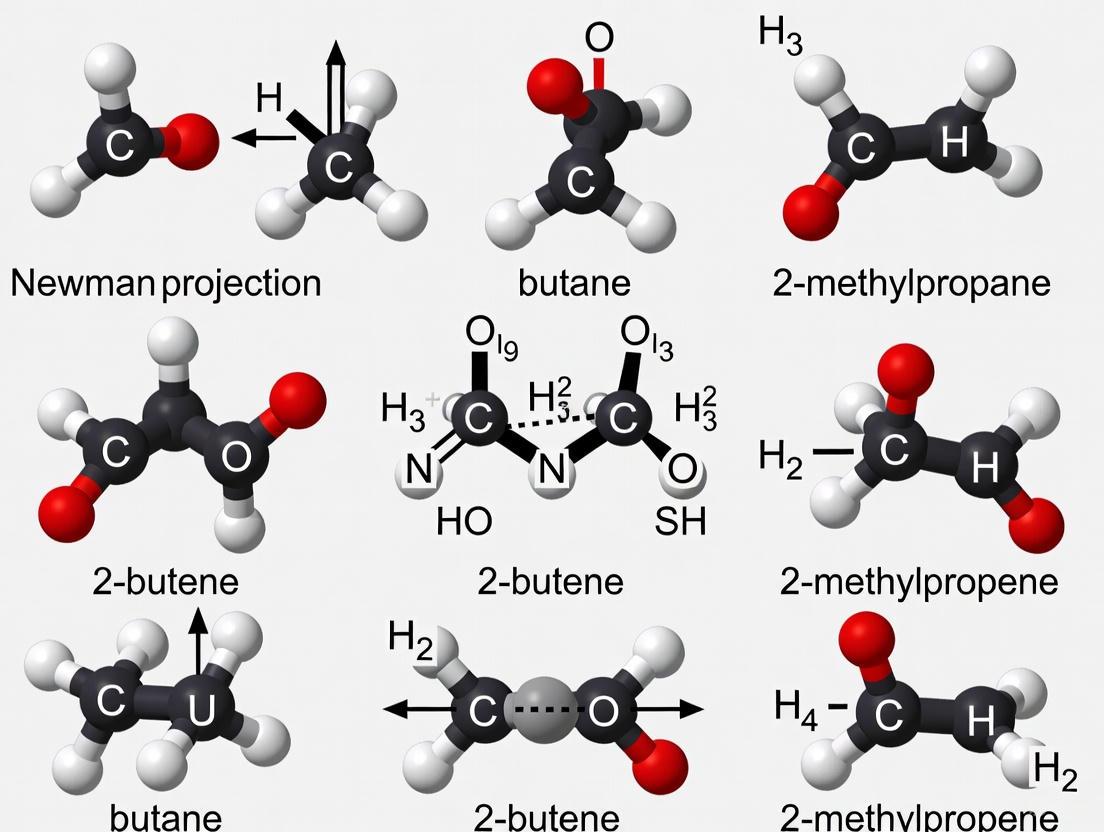
| Cis-Trans Isomers | A subtype of diastereomerism in alkenes or rings, where groups are on the same (cis) or opposite (trans) sides [2]. | Cis isomers often have higher boiling points and lower melting points than their trans counterparts [2]. | cis- and trans-2-butene [2]. |
Chiral molecules are optically active, meaning they rotate the plane of plane-polarized light. One enantiomer rotates light clockwise (dextrorotatory, labeled (+)) and the other counterclockwise (levorotatory, labeled (–)) [3]. The absolute configuration of a chiral center is unambiguously described using the Cahn-Ingold-Prelog (CIP) system, which assigns an ( R ) (from Latin rectus) or ( S ) (from Latin sinister) descriptor based on the atomic numbers of the substituents [3]. For sugars and amino acids, an older but still prevalent D/L system is used, which references the absolute configuration of D- or L-glyceraldehyde [5].
When a chiral compound is synthesized in the lab without a chiral influence, it typically forms a racemic mixture (or racemate), a 50:50 mixture of both enantiomers [3]. In a chiral environment, such as the human body, enantiomers can behave as entirely different substances. For instance, the D-enantiomer of the drug isoproterenol is effective for treating heart rate issues, whereas the L-enantiomer acts on blood pressure [4].
Analytical Methods for Determining Stereochemistry
Determining the precise three-dimensional structure of a molecule is a critical step in research. The methodologies can be broadly categorized into physical, spectroscopic, and chemical techniques, each providing complementary information.
Physical and Spectroscopic Methods
Table 2: Key Analytical Methods for Stereochemical Determination
| Method | Underlying Principle | Application in Stereochemistry |
|---|---|---|
| Polarimetry | Measures the rotation of plane-polarized light by a chiral substance [2]. | Determines optical activity and enantiomeric purity; used to quantify the specific rotation ([α]) of a sample. |
| X-ray Crystallography | Uses X-ray diffraction patterns from a crystallized sample to determine electron density [2]. | Provides the most direct and unambiguous determination of absolute configuration and the 3D arrangement of all atoms in a molecule. |
| Nuclear Magnetic Resonance (NMR) | Exploits the magnetic properties of atomic nuclei in a magnetic field [2]. | 1H/13C NMR: Reveals connectivity and chemical environment.NOE (Nuclear Overhauser Effect): Measures through-space interactions to determine relative configuration and conformation [2]. |
| Chiral Derivatization | Converts enantiomers into diastereomers by reacting with a single-enantiomer chiral reagent [2]. | Allows for the separation and analysis of enantiomers using standard achiral methods like HPLC or GC, as diastereomers have different physical properties. |
The experimental workflow for structural elucidation often integrates multiple techniques, as shown in the following protocol.
Experimental Protocol: Determining Enantiomeric Excess (e.e.) via Chiral HPLC
Objective: To determine the enantiomeric purity of a chiral synthesis product.
- Sample Preparation: Dissolve a small quantity (∼1-5 mg) of the synthesized chiral compound in a suitable solvent (e.g., hexane/isopropanol mixture) to a concentration of approximately 1 mg/mL. Filter the solution through a 0.45 μm syringe filter to remove particulates.
- Chiral HPLC Setup:
- Column: Install a dedicated chiral stationary phase column (e.g., amylose- or cellulose-based).
- Mobile Phase: Prepare an isocratic or gradient eluent, often a mixture of alkane and alcohol (e.g., 90:10 Hexane:Isopropanol).
- Conditions: Set flow rate (e.g., 1.0 mL/min), column temperature (e.g., 25 °C), and detection wavelength (e.g., UV-Vis at 254 nm).
- Calibration and Analysis:
- First, inject the racemic mixture (if available) to establish retention times for both enantiomers and ensure baseline resolution.
- Inject the synthesized sample.
- Data Analysis: Integrate the peak areas for each enantiomer. Calculate the enantiomeric excess (e.e.) using the formula: [ e.e. (\% ) = \frac{| [R] - [S] |}{[R] + [S]} \times 100 = \frac{| AreaR - AreaS |}{AreaR + AreaS} \times 100 ] A high e.e. indicates a highly enantioselective synthesis.
Quantitative Models and Machine Learning in Stereochemistry
Moving beyond qualitative understanding, quantitative models are essential for predicting stereochemical outcomes. A prominent example is the statistical mechanical model used to quantify stereochemical communication in metal-organic assemblies [6]. This model treats each metal center in a self-assembled cage as a two-state system (Δ or Λ configuration). It introduces two key parameters:
- Intra-vertex coupling ((f_1)): The free energy penalty for incorporating an "incorrect" chiral amine enantiomer at a single metal center, quantifying the local chiral induction strength.
- Inter-vertex coupling ((f_2)): The free energy associated with a ligand connecting metal centers with opposite configurations, quantifying the propagation of stereochemical information across the assembly [6].
By fitting experimental data from "sergeant-and-soldiers" experiments to this model, researchers can extract (f1) and (f2) values, providing a unified understanding of how factors like metal ion identity, ligand length, and chiral residue structure influence the overall stereochemistry of complex supramolecular systems [6].
More recently, machine learning (ML) has emerged as a powerful tool for quantitatively predicting stereoselectivity, a task traditionally difficult for computational methods. A composite ML approach has been developed to predict enantioselectivity ((\Delta \Delta G^\ddag)) for chiral phosphoric acid (CPA)-catalyzed reactions [4].
Table 3: Machine Learning Models for Predicting Enantioselectivity
| ML Algorithm | Application Example | Key Advantage |
|---|---|---|
| Random Forest (RF) | Prediction of enantioselectivity in glycosylation and asymmetric catalytic reactions [4]. | Handles non-linear relationships and interactions between molecular features. |
| Support Vector Regression (SVR) | Prediction of enantioselectivities in thiol addition to N-acylimines [4]. | Effective in high-dimensional spaces. |
| LASSO Regression | Feature selection and prediction for asymmetric phenolic dearomatization [4]. | Performs variable selection and regularization to enhance prediction accuracy. |
| Deep Neural Networks (DNN) | Predicting enantioselectivity of catalytic asymmetric β-C-H bond activation [4]. | Capable of learning complex, hierarchical patterns from large datasets. |
The ML workflow involves training models on datasets comprising hundreds of reactions, with features derived from density functional theory (DFT) calculations and molecular topologies describing the catalyst, solvent, nucleophile, and imine components [4]. The composite method uses Bayesian optimization for hyperparameter tuning and a Gaussian Mixture Model (GMM) to cluster new reactions and assign the most appropriate pre-trained ML model for accurate prediction of (\Delta \Delta G^\ddag) [4].
Applications in Drug Discovery and Development
The impact of stereochemistry is perhaps most acutely felt in the pharmaceutical industry, where the principle that "enantiomers should be considered different drugs" is a guiding tenet [3]. Approximately 50% of marketed drugs are chiral, and of these, about half were initially sold as racemic mixtures [3].
Single-Enantiomer Drugs offer several potential advantages over racemates:
- Improved Pharmacologic Profile: Often, the therapeutic activity resides predominantly in one enantiomer (the eutomer), while the other (the distomer) may be inactive or contribute to side effects [3] [7].
- Simpler Pharmacokinetics: Enantiomers can be metabolized at different rates, leading to complex pharmacokinetics for a racemate. Using a single enantiomer simplifies the dose-response relationship [3].
- Reduced Drug Interactions: The distomer may inhibit enzymes or interact with off-target receptors, leading to unwanted drug interactions [3].
A classic case is the antidepressant citalopram and its single-enantiomer derivative escitalopram. Citalopram is a racemic mixture, but the (S)-enantiomer (escitalopram) is responsible for the serotonin reuptake inhibition. The (R)-enantiomer is not only less active but may counteract the therapeutic effects of the (S)-enantiomer. Clinically, 10 mg of escitalopram was shown to be as effective as 40 mg of the racemic citalopram, demonstrating the profound therapeutic advantage of the single-enantiomer formulation [7].
Regulatory agencies like the FDA and EMA require strict control over stereochemistry. Sponsors must identify the stereochemical composition of a drug substance, develop chiral analytical methods early, and justify the choice of developing a racemate versus a single enantiomer [7]. This has led to the strategy of "chiral switching," where a company develops a single-enantiomer version of a previously racemic drug, as seen with esomeprazole (Nexium) from omeprazole (Prilosec) [7].
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for Stereochemical Studies
| Reagent / Material | Function | Application Example |
|---|---|---|
| Chiral Stationary Phase (CSP) HPLC Columns | To separate and analyze enantiomers based on their differential interaction with a chiral solid phase [7]. | Determining enantiomeric purity (e.e.) of synthesis products; analytical and preparative separation. |
| Chiral Solvating Agents (CSAs) | To form transient diastereomeric complexes with enantiomers in solution, making them distinguishable by NMR [2]. | NMR analysis for enantiomeric composition and absolute configuration assignment. |
| Chiral Catalysts (e.g., Chiral Phosphoric Acids - CPAs) | To catalyze reactions enantioselectively, favoring the formation of one enantiomer over the other [4]. | Asymmetric synthesis, such as the addition of nucleophiles to imines to create chiral centers with high e.e. |
| Chiral Derivatizing Agents | To covalently attach a chiral moiety to enantiomers, converting them into diastereomers with different physical properties [2]. | Enabling separation of enantiomers on achiral chromatographic systems or analysis by other methods. |
| Enantiopure Building Blocks (e.g., D-/L- amino acids, sugars) | Serve as chiral starting materials or templates with a known, defined absolute configuration [5]. | Synthesis of complex chiral molecules, peptides, and natural products in a stereocontrolled manner. |
Stereochemistry is a foundational pillar of modern chemical science, inextricably linking the three-dimensional architecture of a molecule to its function. For researchers and drug development professionals, a deep and practical understanding of stereochemical principles is non-negotiable. It is critical for interpreting spectroscopic data, designing synthetic routes, predicting biological activity, and ensuring the safety and efficacy of pharmaceutical agents. The field continues to evolve, with advanced quantitative models and machine learning approaches now providing powerful tools to predict and rationalize stereoselectivity, moving beyond traditional trial-and-error methods. As research delves deeper into complex molecular systems, from supramolecular cages to novel energetic materials, the principles of stereochemistry will remain central to innovation and discovery.
Isomerism constitutes a foundational concept in organic chemistry and a critical consideration in modern drug discovery and development. It describes the phenomenon whereby molecules share the same molecular formula but differ in the arrangement of their atoms [8]. For researchers and drug development professionals, a nuanced understanding of isomerism is not merely academic; it directly impacts the physicochemical properties, biological activity, metabolic fate, and ultimate therapeutic efficacy of molecular entities [9]. The broadest classification divides isomers into two primary categories: structural isomers (also known as constitutional isomers) and stereoisomers [10] [11]. This classification is paramount because the "type" of isomerism dictates the strategies required for synthesis, separation, analysis, and purification—processes central to pharmaceutical development [9].
The significance of this field is powerfully illustrated by historical lessons. The case of thalidomide, a drug administered as a racemic mixture in the late 1950s, remains a stark reminder. While one enantiomer provided the desired sedative effect, its mirror image isomer caused severe teratogenic effects [12]. This tragedy underscored the critical importance of stereochemistry in pharmacology and catalyzed a paradigm shift in how regulatory agencies evaluate chiral drugs, compelling the industry to develop sophisticated methods for isomer-specific synthesis and analysis [12]. Within the context of a broader thesis on stereochemistry, this guide provides a comprehensive technical framework for classifying isomers, complete with experimental protocols essential for research and development.
Foundational Concepts and Classification Framework
At its core, isomerism recognizes that a single molecular formula can correspond to multiple, distinct chemical entities. The fundamental dichotomy in isomer classification rests on the nature of the difference between these entities.
Structural Isomers (Constitutional Isomers) are defined by a different connectivity of atoms [8] [13]. They possess the same molecular formula but differ in the sequence in which their atoms are bonded together. This different bonding architecture inherently means that structural isomers are different compounds with unique IUPAC names and often vastly different chemical and physical properties [11].
Stereoisomers, by contrast, share the same atomic connectivity (the same structural formula) but differ in the three-dimensional orientation of their atoms in space [14]. This category includes a range of isomers from enantiomers to conformational isomers, all of which are detailed in the sections that follow.
The following diagram illustrates the logical decision tree for classifying isomers, providing a roadmap for researchers to categorize any given pair of molecules.
Figure 1: Logical workflow for isomer classification.
Structural Isomerism
Types and Examples
Structural isomerism arises when atoms are bonded in a fundamentally different sequence. The different connectivities lead to distinct skeletal frameworks and/or functional groups. The table below summarizes the primary types of structural isomerism, which include chain, position, and functional group isomerism [8] [15].
Table 1: Classification and Examples of Structural Isomerism
| Type of Structural Isomerism | Defining Principle | Example Molecular Formula | Example Isomers |
|---|---|---|---|
| Chain Isomerism [8] [13] | Different arrangements of the carbon skeleton (e.g., straight chain vs. branched) | C4H10 | Butane (straight-chain), 2-Methylpropane (branched) |
| Position Isomerism [8] [15] | Same functional group or substituent located at different positions on the same carbon skeleton | C5H11Br | 1-Bromopentane, 2-Bromopentane, 3-Bromopentane |
| Functional Group Isomerism [8] [15] | Different functional groups, leading to compounds from different homologous series | C3H6O | Propanal (aldehyde), Propanone (ketone) |
Experimental Protocols for Separation and Analysis
The separation of structural isomers is typically feasible through standard chemical methods because their different connectivities impart significant differences in physical properties like boiling point, melting point, and polarity.
Protocol 1: Fractional Distillation for Chain Isomers
- Principle: Leverages differences in boiling points (bp) arising from variations in molecular surface area and branching. Linear isomers typically have higher boiling points than branched isomers due to stronger London dispersion forces [8].
- Methodology: The isomeric mixture is heated in a distillation apparatus. For example, to separate the chain isomers of C5H12, the mixture is carefully heated. n-Pentane (bp ~36°C) distills over first, followed by isopentane (bp ~28°C), and finally neopentane (bp ~10°C) [8]. The fractions are collected separately based on their predetermined boiling point ranges.
- Key Analysis: Purity of collected fractions is confirmed by Gas Chromatography (GC), comparing retention times against authentic samples.
Protocol 2: Chromatographic Separation for Positional Isomers
- Principle: Utilizes differences in polarity and adsorption affinity. For instance, 1-Bromopentane is slightly less polar than 2-Bromopentane due to the position of the polar bromine atom relative to the alkyl chain [15].
- Methodology: Column Chromatography. The isomeric mixture is applied to a silica gel column. A non-polar mobile phase (e.g., hexane) is used initially, gradually increasing polarity with a more polar solvent (e.g., ethyl acetate). The less polar isomer elutes first.
- Key Analysis: Fractions are collected and analyzed by Thin-Layer Chromatography (TLC). Further structural confirmation is achieved using Nuclear Magnetic Resonance (NMR) spectroscopy, where the chemical shifts and splitting patterns of protons near the functional group are diagnostically different [13].
Stereoisomerism
Stereoisomers possess an identical bond connectivity but a different spatial arrangement of atoms. This broad category is first divided into configurational isomers and conformational isomers. Configurational isomers are stereoisomers that cannot be interconverted readily because the process requires breaking and reforming bonds (e.g., enantiomers, diastereomers, cis-trans isomers) [14]. Conformational isomers, on the other hand, can be interconverted rapidly by rotation around single bonds (e.g., the chair and boat forms of cyclohexane) [14] [16].
Table 2: Classification of Stereoisomers with Defining Features
| Category | Subtype | Key Defining Feature | Example | Separable at RT? |
|---|---|---|---|---|
| Configurational Isomers | Enantiomers [14] [12] | Non-superimposable mirror images; contain chiral centers. | D- & L-lactic acid [15] | Yes |
| Diastereomers [14] [11] | Stereoisomers that are not mirror images. | Cis- & Trans-1,2-dimethylcyclopropane [16] | Yes | |
| Geometric (Cis-Trans/E-Z) [14] [10] | Differ in arrangement about a double bond or ring due to restricted rotation. | Cis- & Trans-2-butene [10] | Yes | |
| Conformational Isomers | Rotamers (e.g., Staggered, Eclipsed) [16] | Differ by rotation around a single bond. | Staggered & Eclipsed ethane | No |
| Ring Conformers [16] | Different puckering modes of a ring system. | Chair & Boat cyclohexane | No |
Enantiomers and Chirality
Enantiomers are a pair of stereoisomers that are non-superimposable mirror images of each other [12] [16]. A molecule that is not superimposable on its mirror image is described as chiral. The most common source of chirality is a chiral center, typically a carbon atom bonded to four different substituents [12].
- Optical Activity: The most defining property of enantiomers is their ability to rotate the plane of plane-polarized light. One enantiomer will rotate the light in a clockwise direction (dextrorotatory, labeled as (+) or d), while the other will rotate it counterclockwise (levorotatory, labeled as (-) or l) by an equal magnitude [12].
- Physical and Biological Properties: Enantiomers have identical physical properties (melting point, boiling point, solubility) in an achiral environment. However, their interactions with other chiral molecules, such as biological receptors and enzymes, can be dramatically different, leading to distinct pharmacological effects, as seen with thalidomide [12].
Diastereomers, Geometric Isomers, and Conformers
- Diastereomers: These are stereoisomers that are not mirror images [14] [11]. Unlike enantiomers, diastereomers have different physical properties (e.g., melting points, solubilities) and can therefore be separated by conventional techniques like fractional crystallization or chromatography [12].
- Geometric Isomers (Cis-Trans / E-Z): This form of diastereomerism arises from restricted rotation around a double bond or in a ring system [14] [10]. In the E/Z system (used for more complex alkenes), priority is assigned to substituents based on atomic number. If the two highest-priority groups are on the same side, it is the Z-isomer (zusammen, together); if on opposite sides, it is the E-isomer (entgegen, opposite) [14].
- Conformational Isomers: These represent different spatial arrangements of atoms achieved by rotation around single bonds [14] [16]. In cyclohexane, the chair conformation is the most stable energy minimum, while the boat conformation represents a higher-energy form. Despite being rapidly interconverting, different conformers can have profoundly different steric and electronic environments, influencing reactivity.
Experimental Protocols for Stereoisomer Analysis
Protocol 3: Polarimetry for Enantiomer Characterization
- Principle: Measures the angle and direction by which a chiral compound rotates plane-polarized light [12].
- Methodology: A solution of the pure chiral compound is prepared in a suitable solvent at a known concentration (c, in g/mL) and placed into a sample cell of specific path length (l, in dm). The polarimeter is zeroed with the pure solvent. The sample is inserted, and the observed rotation (α) is measured. The specific rotation [α] is calculated as: [α] = α / (l * c).
- Key Analysis: The specific rotation is a physical constant used to identify and characterize enantiomers. A racemic mixture (a 50:50 mixture of enantiomers) will show no net optical rotation [12].
Protocol 4: Chiral Chromatography for Enantiomer Separation
- Principle: Uses a chromatographic column with a chiral stationary phase (CSP) that interacts differentially with each enantiomer, creating a diastereomeric interaction complex and allowing separation [9].
- Methodology: The racemic mixture is injected into a High-Performance Liquid Chromatography (HPLC) system equipped with a CSP (e.g., cyclodextrin, macrocyclic glycopeptide). An achiral mobile phase is used. The two enantiomers, interacting differently with the CSP, elute at different retention times (tR1 and tR2).
- Key Analysis: The resolution (Rs) between the two peaks is calculated. The identity of each peak can be confirmed by comparison with the known specific rotation of the collected fractions or by co-injection with a pure enantiomer standard.
The following diagram visualizes the key instrumental workflow for separating and analyzing stereoisomers, integrating chiral chromatography and polarimetry.
Figure 2: Workflow for chiral separation and analysis.
The Research Toolkit: Essential Reagents and Materials for Isomer Research
The following table details key reagents, materials, and instruments essential for experimental work in isomer synthesis, separation, and analysis.
Table 3: Essential Research Reagents and Materials for Isomer Research
| Reagent/Material/Instrument | Primary Function in Isomer Research |
|---|---|
| Silica Gel (for Column Chromatography) [13] | A polar stationary phase for separating structural isomers and diastereomers based on adsorption affinity differences. |
| Chiral HPLC Columns [9] | Columns with a chiral stationary phase (CSP) for the analytical and preparative separation of enantiomers. |
| Polarimeter [12] | Instrument for measuring the optical activity of chiral compounds, used to determine enantiomeric purity and identity. |
| Chiral Derivatizing Agent (e.g., MTPA-Cl) | A chiral reagent that reacts with a racemic mixture to form a pair of diastereomers, which can then be separated using standard achiral methods (e.g., silica gel chromatography). |
| Deuterated Solvents (for NMR) | Essential solvents for NMR spectroscopy, used to confirm molecular structure, identify isomer type, and determine enantiomeric purity using chiral shift reagents. |
| Gas Chromatograph (GC) | Instrument for separating and analyzing volatile mixtures of isomers, particularly effective for chain and positional isomers. |
The rigorous classification of isomers into structural and stereoisomers provides an indispensable framework for scientific research, particularly in the pharmaceutical industry. The ability to distinguish, separate, and characterize these molecular variants is not a mere academic exercise but a fundamental requirement for developing safe and effective therapeutics. As the field advances, the focus on isomer-specific drug formulations is intensifying, driven by the demands of personalized medicine and the lessons of history [9]. The experimental protocols and analytical techniques outlined in this guide form the bedrock of this endeavor. Future research will continue to leverage advancements in nanotechnology, computational modeling, and synthetic methodology to achieve ever-greater control over molecular geometry, pushing the boundaries of drug delivery and therapeutic efficacy [17] [9]. A deep and practical understanding of isomerism, as detailed in this comprehensive guide, remains a cornerstone of innovation in chemistry and drug development.
Chirality, derived from the Greek word for "hand," describes the fundamental geometric property of a molecule that is non-superimposable on its mirror image [18]. This "handedness" in molecular architecture is not merely an abstract chemical concept but a critical determinant of biological activity, particularly in pharmaceutical science. The three-dimensional spatial arrangement of atoms around specific stereogenic elements dictates how a drug molecule interacts with its biological targets, which are themselves chiral entities such as proteins, enzymes, and receptors [19] [18]. The historical recognition of this phenomenon dates back to Louis Pasteur's 1848 manual separation of tartaric acid crystals, which established the foundational principles of molecular asymmetry [19] [18].
In pharmaceutical contexts, the distinction between enantiomers—non-superimposable mirror image molecules—can mean the difference between therapeutic benefit and detrimental toxicity. The tragic case of thalidomide, where one enantiomer provided desired sedative effects while its mirror image caused severe birth defects, starkly illustrates the life-or-death implications of stereochemistry in drug development [18]. Modern regulatory agencies now require rigorous stereochemical evaluation of new drug candidates, recognizing that enantiomers can exhibit marked differences in pharmacology, toxicology, pharmacokinetics, and metabolism [19]. Contemporary research indicates that over half of all marketed drugs are chiral compounds, with approximately 90% of these historically marketed as racemic mixtures (equimolar combinations of both enantiomers), though current practice increasingly favors development of single-enantiomer drugs [19].
Stereogenic Elements: Beyond the Carbon Center
Classical Stereogenic Centers
Traditional stereochemistry has primarily focused on carbon-centered chirality, where a tetrahedral carbon atom bears four distinct substituents [18]. This asymmetric carbon center represents the most prevalent stereogenic element in organic molecules and pharmaceuticals. The configuration around such centers is conventionally described using the Cahn-Ingold-Prelog (CIP) priority rules, which assign descriptors of R (rectus) or S (sinister) based on the atomic numbers and masses of substituents systematically arranged in three-dimensional space [19]. In molecular representations, wedge-dash notation depicts three-dimensional orientation with solid wedges indicating bonds projecting toward the viewer and hashed wedges representing bonds receding away from the viewer [18]. For molecules with multiple chiral centers, Fischer projections provide a two-dimensional schematic representation where horizontal lines indicate bonds projecting outward from the plane and vertical lines represent bonds extending behind the plane [18].
Table 1: Classical and Emerging Stereogenic Elements in Pharmaceutical Compounds
| Stereogenic Element Type | Structural Basis | Configuration Descriptors | Pharmaceutical Examples |
|---|---|---|---|
| Carbon center | Tetrahedral carbon with four different substituents | R/S | (S)-Propranolol, (S)-Naproxen |
| Nitrogen center | Tertiary amines in rigid skeletons | N-inversion limited | Quinine derivatives |
| Chiral axis | Restricted rotation around single bonds | P/M (or R/S) | Atropisomeric drugs |
| Chiral plane | Helical or planar structures with hindered rotation | N/A | Helicenes |
| All-heteroatom spiro center | Carbon with four heteroatom substituents (O, N) | R/S | UNIGE experimental molecules |
Emerging Heteroatomic Stereogenic Elements
While carbon-centered chirality has been extensively studied, recent research has revealed the substantial importance and unique challenges of heteroatomic stereogenic elements. Nitrogen-centered chirality has proven particularly challenging to control due to the relatively low inversion barrier between nitrogen enantiomers, which typically leads to rapid racemization [20]. Strategies to stabilize chiral nitrogen centers include incorporation into quaternary ammonium salts, amine N-oxides, metal coordination complexes, or rigid polycyclic skeletons as found in natural products like quinine [20]. Boron-centered chirality has also emerged as a significant frontier, particularly in pharmaceutical and materials chemistry [20].
A groundbreaking advance comes from researchers at the University of Geneva (UNIGE) and University of Pisa, who have designed a novel family of chiral molecules featuring an unprecedented all-heteroatom-substituted carbon spiro stereocenter [21] [22]. Unlike traditional chiral centers where carbon is bound to carbon-based substituents, these innovative structures feature a central carbon atom bonded exclusively to oxygen and nitrogen atoms [21]. This architectural approach represents "a major conceptual and experimental breakthrough" in stereochemistry, marking the first isolation of such molecules in stable form [21] [22].
The most remarkable property of these novel chiral molecules is their exceptional configurational stability. Using dynamic chromatography and quantum chemistry calculations, researchers demonstrated that the half-life for racemization (conversion from one enantiomer to its mirror image) reaches approximately 84,000 years at room temperature for one molecule, and 227 days at 25°C for another variant [21] [22]. This extraordinary stability—essentially creating 'mirror-proof' molecular architectures—has profound implications for pharmaceutical development, as it guarantees drug integrity without requiring specialized storage conditions and prevents the transformation of therapeutic enantiomers into potentially harmful counterparts over time [21].
Quantitative Analysis of Chiral Stability and Drug Properties
The biological implications of molecular chirality are quantifiable through numerous pharmacokinetic and pharmacodynamic parameters. Enantioselective interactions with biological systems can produce dramatically different dose-response relationships, metabolic profiles, and toxicity thresholds between enantiomers of the same compound.
Table 2: Enantioselective Pharmacological Properties of Representative Chiral Drugs
| Drug Compound | Therapeutic Activity by Enantiomer | Potency Ratio (Eutomer:Distomer) | Clinical Implications |
|---|---|---|---|
| Propranolol | S(-): β-adrenergic blockadeR(+): Minimal β-blockadeR(+): Inhibits T4 to T3 conversion | 100:1 (β-blockade) | Racemate: contraindicated in thyroid disordersSingle R-enantiomer: potential for hyperthyroidism |
| Methadone | (R)-methadone: Opioid analgesia(S)-methadone: hERG binding, cardiac risk | N/A | (S)-enantiomer associated with QT prolongation and cardiac arrest |
| β-blockers (class) | S(-): β-adrenoceptor blockade | Varies by compound | Most marketed as racemates except timolol, penbutolol |
| Amine boranes (experimental) | Continuous N-B stereocenters | High diastereo-/enantioselectivity | Potential chiral transfer hydrogenation reagents |
Advanced Methodologies in Stereochemical Analysis and Control
Experimental Protocols for Stereochemical Construction
The precise construction of complex stereogenic architectures requires sophisticated synthetic methodologies. Recent advances in copper-catalyzed asymmetric B-H insertion reactions demonstrate state-of-the-art protocols for creating challenging continuous stereogenic centers incorporating nitrogen and boron atoms [20].
Protocol: Copper-Catalyzed Asymmetric B-H Insertion for Continuous Stereogenic Centers
- Reaction Setup: In a flame-dried Schlenk flask under inert atmosphere, combine racemic cyclic amine borane (1a, 1.0 equiv) and diaryl diazomethane (2a, 1.2 equiv) in anhydrous dichloromethane (DCM) as solvent [20].
- Catalyst System: Employ copper(I) thiophene-2-carboxylate hydrate (CuTc, 5 mol%) with specialized bisoxazoline (BOX) ligand L1 (20 mol%) as the chiral controller [20]. The additive potassium tetrakis(perfluorophenyl)borate (KBArF, 20 mol%) is crucial for enhancing reaction efficiency [20].
- Reaction Conditions: Conduct the transformation at -70°C with strict temperature control to maintain high enantioselectivity. Reaction monitoring via TLC or in situ spectroscopy is recommended [20].
- Workup and Isolation: Upon completion, quench the reaction with saturated aqueous ammonium chloride solution. Extract with DCM (3 × 15 mL), dry the combined organic layers over anhydrous magnesium sulfate, filter, and concentrate under reduced pressure [20].
- Purification and Analysis: Purify the crude product by flash chromatography on silica gel. Analyze enantiomeric purity by chiral HPLC or SFC, and determine absolute configuration by X-ray crystallography or computational methods (e.g., DFT calculations) [20].
This protocol achieves excellent diastereoselectivity (ranging from 4.8:1 to >20:1 dr) and enantioselectivity (84-98% ee) across a broad substrate scope, including electronically varied diazo compounds and substituted amine boranes [20]. The methodology features a meaningful kinetic resolution pathway during the transformation, enabling access to enantiopure boron-coordinated nitrogen-centered compounds that serve as potential chiral transfer hydrogenation reagents [20].
Figure 1: Experimental Workflow for Constructing Continuous Stereogenic Centers via Asymmetric B-H Insertion
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Advanced Stereochemical Research
| Reagent/Catalyst | Function in Stereochemical Research | Application Examples |
|---|---|---|
| Chiral BOX Ligands (L1-L6) | Induce asymmetry in metal-catalyzed reactions | Copper-catalyzed B-H insertion [20] |
| Copper(I) Thiophene-2-carboxylate (CuTc) | Catalytic activation of B-H bonds | Stereoselective transformations [20] |
| KBArF (Potassium tetrakis(perfluorophenyl)borate) | Enhances reaction efficiency as additive | B-H insertion optimization [20] |
| Diazo Compounds | Carbene precursors for insertion reactions | Construction of C-B bonds [20] |
| Amine Boranes | Provide N-B stereogenic elements | Continuous stereocenter formation [20] |
| Chiral Stationary Phases | Enantiomer separation in chromatography | Analysis of enantiomeric purity [21] |
Computational and Machine Learning Approaches
Modern stereochemical research increasingly incorporates computational methods and artificial intelligence to predict, analyze, and optimize chiral molecules. Stereochemistry-aware generative models represent a cutting-edge approach in computational drug discovery that explicitly accounts for three-dimensional molecular arrangement during the design process [23]. These models utilize string-based molecular representations such as SMILES (Simplified Molecular-Input Line-Entry System), SELFIES (SELF-Referencing Embedded Strings), and GroupSELFIES, which natively encode stereochemical information through specialized tokens denoting R/S and E/Z configurations [23].
Benchmarking studies demonstrate that stereochemistry-aware models perform comparably to or surpass conventional algorithms in stereochemistry-sensitive tasks, including optimization of binding affinity, metabolic stability, and optical activity [23]. However, researchers must consider the trade-off between stereochemical precision and chemical space complexity, as stereo-aware models navigate an expanded molecular landscape that includes all possible stereoisomers [23]. These computational approaches are particularly valuable for predicting the biological activity of enantiomers and avoiding problematic stereochemical configurations, such as the (S)-methadone enantiomer associated with hERG channel binding and cardiotoxicity [23].
Figure 2: Computational Workflow for Stereochemistry-Aware Molecular Generation
Future Perspectives and Research Directions
The field of stereochemistry in drug molecules continues to evolve with several promising research frontiers. Catalytic deracemization strategies represent a paradigm shift in accessing single enantiomers from racemic mixtures without traditional separation techniques. The CALIDE (Catalytic Light-induced Deracemization) project explores photochemical approaches that temporarily erase stereogenic information and recreate it using light as the exclusive energy source, potentially establishing "one of the key pillars on which the future preparation of enantiomerically pure compounds will rest" [24].
The development of continuous stereogenic frameworks incorporating multiple heteroatoms (N-B, N-B-C) expands the toolbox for designing molecular architectures with precisely controlled three-dimensional shapes [20]. These complex chiral environments enable fine-tuning of drug-target interactions that may lead to improved specificity and reduced off-target effects. Additionally, the integration of stereochemistry-aware machine learning models into drug discovery pipelines promises to accelerate the identification of optimal chiral configurations while avoiding problematic stereochemical features associated with toxicity or metabolic instability [23].
As these technologies mature, the pharmaceutical industry is poised to transition from primarily carbon-centered chirality to embrace diverse stereogenic elements including stable heteroatomic centers, axial chirality, and helical structures. This expansion of the stereochemical lexicon will provide medicinal chemists with unprecedented control over molecular geometry, ultimately enabling the design of safer, more effective therapeutic agents with precisely optimized three-dimensional architectures.
Stereochemistry, the study of the three-dimensional arrangement of atoms in molecules, is a fundamental concept with profound implications in organic chemistry and biological research. A molecule's spatial orientation directly dictates its interactions with biological systems, making stereochemistry not merely an academic exercise but a critical consideration in fields such as drug discovery and development [25]. Isomers—molecules with identical molecular formulas but different atom arrangements—are broadly categorized into constitutional isomers (differing in bond connectivity) and stereoisomers (identical connectivity but different spatial arrangement) [11] [26]. Stereoisomers are further subdivided into enantiomers and diastereomers, two classes that exhibit distinct properties and biological behaviors. Within the chiral environment of biological systems, these differences can dictate efficacy, toxicity, and the overall fate of a molecule, underscoring the necessity for researchers to adeptly identify and separate these isomers [27] [28].
Fundamental Concepts and Definitions
Stereoisomers: Enantiomers and Diastereomers
Stereoisomers share the same atomic connectivity but differ in the orientation of their atoms in three-dimensional space [11]. This difference arises from the presence of stereogenic centers, most commonly chiral carbon atoms bonded to four different substituents [3].
- Enantiomers are pairs of stereoisomers that are non-superimposable mirror images of each other [29] [30]. This relationship requires that all chiral centers in the molecule have opposite configurations. A molecule with one chiral center exists as a pair of enantiomers.
- Diastereomers are stereoisomers that are not mirror images of each other [29] [30]. This occurs when molecules have multiple chiral centers and the configurations differ at one or more, but not all, of these centers [30]. Unlike enantiomers, which always exist in pairs, a single molecule can have multiple diastereomers.
Table 1: Core Definitions and Relationships
| Feature | Enantiomers | Diastereomers |
|---|---|---|
| Mirror Image Relationship | Non-superimposable mirror images [30] [31] | Non-mirror images [30] [31] |
| Configuration at Chiral Centers | Opposite configuration at every chiral center [30] | Different configuration at one or more, but not all chiral centers [30] |
| Number of Possible Isomers | Always a pair | Can be several molecules for a compound with multiple chiral centers [31] |
Key Differentiating Workflow
The following diagram outlines the logical decision process for classifying the relationship between two stereoisomers.
Comparative Properties of Enantiomers and Diastereomers
The distinct spatial relationships between enantiomers and diastereomers manifest in significantly different physical, chemical, and biological properties.
Physical and Chemical Properties
Table 2: Comparative Physical and Chemical Properties
| Property | Enantiomers | Diastereomers |
|---|---|---|
| Physical Properties | Identical in achiral environments (melting point, boiling point, solubility in achiral solvents) [30] | Distinct (different melting points, boiling points, solubilities) [30] |
| Optical Activity | Rotate plane-polarized light equally but in opposite directions [30] | Have different specific rotations; not necessarily equal and opposite [30] |
| Interaction with Achiral Reagents | Identical chemical behavior [30] | Can exhibit different chemical reactivity [30] |
| Separation Methods | Require chiral environments (e.g., chiral chromatography, chiral resolving agents) [30] | Can often be separated by conventional techniques (e.g., distillation, recrystallization, achiral chromatography) [30] |
Biological and Pharmacological Significance
Biological systems are inherently chiral, composed of building blocks like L-amino acids and D-sugars. This homochirality means that enantiomers, despite their identical physical properties in a test tube, are perceived as completely different entities by biological systems [3]. Enzymes, receptors, and transporters can distinguish between them, leading to dramatically different pharmacological profiles [27].
- Enantioselectivity in Pharmacokinetics and Toxicity: The enantiomers of a drug can exhibit differences in absorption, distribution, metabolism, and excretion (ADME) [27]. For instance, carrier-mediated absorption in the intestine can be enantioselective. Distribution can be influenced by preferential binding to plasma proteins like human serum albumin (HSA) or α1-acid glycoprotein (AGP). Most notably, metabolic enzymes often display high enantioselectivity, leading to different metabolic rates and pathways for each enantiomer [27]. This can result in one enantiomer being efficiently detoxified and excreted while the other is metabolized into a toxic compound [27].
- Diastereomers in Drug Action: Because diastereomers have different physical properties and 3D shapes, they bind to biological targets with distinct affinities and can produce different therapeutic or adverse effects. This makes them effectively different drugs from a pharmacological perspective.
Table 3: Biological and Pharmacological Implications
| Aspect | Enantiomers | Diastereomers |
|---|---|---|
| Binding to Biological Targets | Can have drastically different affinities and effects (e.g., one is active, the other inactive or antagonistic) [3] | Have different physical properties and 3D shapes, leading to different binding affinities and biological activities [30] |
| Metabolism | Often metabolized at different rates and/or by different pathways, potentially producing different metabolites [27] | Behave as distinct chemical entities; are metabolized differently |
| Toxicity Profile | One enantiomer may be responsible for desired effect, while the other causes side effects or toxicity (e.g., Thalidomide) [30] | Toxicity profiles are typically uncorrelated and must be evaluated separately |
| Regulatory Considerations | Requires justification for developing a racemate vs. a single enantiomer; stereochemistry must be defined early [28] | Regarded as distinct molecular entities; each requires full characterization |
Analytical Methods for Differentiation and Characterization
Determining the absolute configuration and purity of stereoisomers is crucial in research and development. The following workflow visualizes a multi-technique approach to stereochemical analysis.
Experimental Protocols for Stereochemical Analysis
1. Determination of Absolute Configuration using Chiroptical Methods
- Objective: To unambiguously assign the absolute configuration (AC) of a novel chiral compound, such as a β-lactam derivative, using a combination of computational chemistry and spectroscopic techniques [32].
- Methodology:
- Sample Preparation: Prepare a high-purity sample of the chiral compound. For solution-based methods like Electronic Circular Dichroism (ECD) and Vibrational Circular Dichroism (VCD), select an appropriate solvent and determine the optimal concentration to achieve a good signal-to-noise ratio.
- Theoretical Calculation:
- Perform a conformational search to identify all low-energy conformers of the molecule.
- Optimize the geometry of these conformers using quantum chemical methods (e.g., Density Functional Theory - DFT).
- Calculate the theoretical ECD and/or VCD spectra for the optimized conformers, applying an appropriate solvation model. The final theoretical spectrum is a Boltzmann-weighted average of the spectra of all significant conformers.
- Experimental Data Acquisition:
- Acquire the experimental ECD and VCD spectra of the compound under conditions as close as possible to the theoretical model.
- Data Analysis and Assignment: Compare the experimental spectra with the calculated theoretical spectra. A strong match between the experimental and calculated spectra for one enantiomer allows for the confident assignment of its absolute configuration [32].
2. Enantioselective Analytical Methods for Pharmacokinetic Studies
- Objective: To accurately monitor and quantify the concentration of individual enantiomers in biological matrices (e.g., plasma, urine) during pharmacokinetic studies [27].
- Methodology:
- Sample Collection and Preparation: Collect biological samples at predetermined time points after drug administration. Pre-treat samples using techniques like protein precipitation, liquid-liquid extraction, or solid-phase extraction to remove interfering matrix components.
- Chiral Chromatography:
- Technique: High-Performance Liquid Chromatography (HPLC) or Ultra-High-Performance Liquid Chromatography (UHPLC).
- Stationary Phase: Use a chiral stationary phase (CSP) designed to interact differentially with the two enantiomers. Common CSPs include cyclodextrins, macrocyclic glycopeptides (e.g., teicoplanin), and polysaccharide derivatives (e.g., cellulose tris-3,5-dimethylphenylcarbamate).
- Mobile Phase: Optimize the composition of the mobile phase (organic solvent, buffer pH, ionic strength) to achieve baseline separation of the enantiomers.
- Detection: Couple the chromatographic system to a sensitive detector, such as a mass spectrometer (MS) or a fluorescence detector, for specific and accurate quantification of each enantiomer at low concentrations in complex biological samples [27].
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for Stereochemical Analysis
| Reagent / Material | Function and Application in Stereochemistry |
|---|---|
| Chiral Stationary Phases (CSPs) | Used in HPLC/UHPLC to physically separate enantiomers based on transient diastereomeric complex formation on the column [27]. |
| Chiral Derivatizing Agents | Achiral reagents that react with enantiomers to form covalently bonded diastereomers, which can then be separated using standard achiral chromatography [27]. |
| Chiral Solvating Agents | Additives that create a chiral environment in the solution, used in NMR spectroscopy to cause chemical shift differences between the enantiomers, allowing for their identification and quantification. |
| Enantiopure Building Blocks | Commercially available chiral synthons (e.g., amino acids, sugars, terpenes) used in asymmetric synthesis to introduce specific chirality into a target molecule. |
The distinction between enantiomers and diastereomers is a cornerstone of stereochemistry with profound practical consequences. While enantiomers are indistinguishable in achiral environments, their divergent interactions with biological systems necessitate rigorous characterization and often separate development as unique pharmaceutical agents. Diastereomers, with their distinct physical properties, are treated as different chemical compounds altogether. The drive towards developing single-enantiomer drugs, known as "chiral switches," underscores the importance of stereochemistry in improving therapeutic efficacy and safety profiles [27] [3]. For researchers, a deep understanding of these concepts, coupled with mastery of modern analytical techniques like chiroptical spectroscopy and enantioselective chromatography, is indispensable for success in drug discovery, natural product chemistry, and the development of new chiral materials.
The field of stereochemistry, which governs the three-dimensional arrangement of atoms within molecules and its profound implications for biological activity, finds its origin in a seminal 1848 experiment conducted by Louis Pasteur. While working with crystals of sodium ammonium tartrate, Pasteur observed that paratartaric acid (now known as racemic acid) consisted of two distinct types of crystals that were nonsuperimposable mirror images of one another [33]. Using tweezers, he meticulously separated these left-handed and right-handed crystals and discovered that their solutions rotated plane-polarized light in equal but opposite directions, while the original mixture was optically inactive [33] [34]. This foundational discovery of molecular chirality (from the Greek cheir, meaning "hand") revealed that molecules with identical chemical compositions could exhibit "handedness," a property Pasteur termed molecular dissymmetry [34] [35]. This whitepaper traces the trajectory from this critical observation to its indispensable role in modern pharmaceutical research and drug development, framing these developments within the broader context of stereochemistry and isomerism in organic molecules research.
Pasteur's Seminal Experiment: Methodology and Discovery
Historical and Scientific Background
In the mid-19th century, the molecular basis for optical activity was unknown. Jean-Baptiste Biot had previously established that certain organic substances, including natural tartaric acid derived from wine production, could rotate plane-polarized light [34] [35]. However, a chemically identical substance, paratartaric or racemic acid, showed no such optical activity, presenting a scientific paradox. The prevailing theory from Eilhard Mitscherlich suggested that the tartrate and paratartrate salts had identical crystalline forms, which Pasteur suspected was incorrect [34].
Detailed Experimental Protocol
Materials and Setup
Research Reagent Solutions & Essential Materials
| Item | Function/Description |
|---|---|
| Sodium Ammonium Tartrate Salt | The key substrate, derived from wine production sediments [33] [36]. |
| Polarimeter | An apparatus, pioneered by Biot, used to measure the rotation of plane-polarized light by a solution [34]. |
| Microscope with Magnifying Lens | Essential for observing the small hemihedral facets on the crystals [34]. |
| Tweezers | Used for the manual separation of the left-handed and right-handed crystals [33]. |
| Crystallization Dish | For the slow crystallization of a concentrated solution of the salt [33]. |
Procedure
- Crystallization: A concentrated solution of sodium ammonium tartrate was prepared and allowed to crystallize slowly at a temperature below 28 °C [34]. Pasteur noted that temperature control was critical, as higher temperatures could lead to a different crystalline form (a racemate) where the hemihedral facets would not appear [34].
- Observation and Hypothesis: Under the microscope, Pasteur observed that the crystals of the paratartrate were not identical. They exhibited small hemihedral facets—tiny faces inclined to the left or to the right—making the crystals mirror images of each other [33] [34]. This was the visual manifestation of molecular dissymmetry.
- Separation: Using tweezers, Pasteur painstakingly separated the crystals into two piles: one of "right-handed" crystals and another of "left-handed" crystals [33].
- Analysis: The crystals from each pile were dissolved in water to create separate solutions. These solutions were then analyzed using a polarimeter.
- Results: The solution of right-handed crystals rotated polarized light to the right (dextrorotatory), while the solution of left-handed crystals rotated light to the left (levorotatory) by an equal magnitude [33] [34]. An equal mixture of the two solutions resulted in no net rotation of light, explaining the inactivity of the original racemic acid.
The logical flow of Pasteur's discovery, from initial observation to the conclusive interpretation of molecular chirality, is summarized in the diagram below.
Interpretation and Significance
Pasteur correctly deduced that the dissymmetry observed in the crystals reflected a fundamental dissymmetry at the molecular level [33] [35]. He postulated that the paratartaric acid was not a pure compound but a 1:1 mixture of two different molecular species with opposite asymmetric arrangements—what we now call enantiomers [33] [36]. This discovery was revolutionary because it suggested that a molecule's properties were determined not only by the type and number of its atoms but also by their spatial arrangement. As noted in contemporary analyses, "Pasteur’s vision was extraordinary, for it was not until 25 years later that his ideas regarding asymmetric carbon atoms were confirmed" by van't Hoff and Le Bel, who proposed the tetrahedral carbon atom [33] [37].
The Bridge to Molecular Pharmacology
The principle of stereochemistry revealed by Pasteur established a fundamental code for molecular pharmacology: biological systems are inherently chiral environments [35]. Receptors, enzymes, and other macromolecular targets are themselves composed of chiral building blocks (e.g., L-amino acids) and thus interact stereospecifically with the molecules that bind to them.
Key Quantitative Examples of Stereochemistry in Drug Action
| Drug/Compound | Enantiomer/Eutomer | Pharmacological Activity | Distomer/Other Enantiomer |
|---|---|---|---|
| Thalidomide [37] | (R)-thalidomide | Sedative | (S)-thalidomide (Teratogenic, causes birth defects) |
| Methadone [23] | (R)-methadone | Opioid agonist (pain relief) | (S)-methadone (Binds hERG, cardiotoxic) |
| Citalopram/Escitalopram [7] | (S)-citalopram (Escitalopram) | Potent SSRI (antidepressant) | (R)-citalopram (~30x weaker, may counteract S-isomer) |
| β-blockers (e.g., Propranolol) [7] | L-enantiomer | Beta-adrenergic blockade | D-enantiomer (Significantly less active) |
| Dopa [7] | L-DOPA | Effective for Parkinson's disease | D-DOPA (Inactive in human enzymes) |
| Tartaric Acid [33] [36] | L-tartaric acid | Naturally occurring dextrorotatory form | D-tartaric acid (Levorotatory form) |
This stereospecificity means that two enantiomers can be perceived by the body as two completely different molecules. A powerful analogy is the "lock-and-key" model, where only one key (the correct enantiomer) fits perfectly into the lock (the biological target) to elicit a response [35]. The other enantiomer may have reduced activity, no activity, or an entirely different and potentially adverse effect. This principle directly challenges the pharmaceutical industry to identify the specific stereochemical isomer responsible for a drug's efficacy and safety [35].
Modern Pharmaceutical Applications and Protocols
Stereochemistry in Drug Discovery and Development
The legacy of Pasteur's discovery is deeply embedded in every stage of modern drug development, from initial screening to regulatory approval.
- Structure-Activity Relationship (SAR): Medicinal chemists treat each stereoisomer as a distinct molecule during SAR exploration [7]. The eudismic ratio (the ratio of activity between the eutomer and distomer) is used to quantify the stereoselectivity of a compound for its target [7].
- Screening Libraries: There is a strategic shift from "flat" aromatic compound libraries to 3D-enriched libraries with higher Fsp3 (fraction of sp3 carbons) and stereogenic centers to improve target specificity and metabolic stability [7] [23]. Strategies involve screening either separate enantiomers or racemic mixtures followed by "deconvolution" to identify the active enantiomer [7].
- Regulatory Landscape: Regulatory bodies (ICH/FDA/EMA) require strict control over stereochemistry [7]. Guidelines mandate the identification of stereochemical composition, development of chiral analytical methods (e.g., chiral HPLC), and justification for developing a racemate versus a single enantiomer. For racemates, the pharmacokinetics and pharmacodynamics of both enantiomers must be characterized [7].
Experimental and Computational Methodologies
Chiral Resolution Protocol
This modern methodology descends directly from Pasteur's initial manual separation.
- Diastereomeric Salt Formation: The racemic mixture of a chiral acid (or base) is reacted with a single enantiomer of a chiral base (or acid) to form a pair of diastereomeric salts [34].
- Fractional Crystallization: These diastereomeric salts have different physical properties (e.g., solubility) and can be separated through fractional crystallization.
- Regeneration: The separated diastereomeric salts are then treated with a strong acid or base to regenerate the purified enantiomers of the original compound.
Computational Drug Design
Modern in silico methods must explicitly account for stereochemistry. The workflow for stereochemistry-aware molecular generation is illustrated below.
- String-Based Representations: Simplified Molecular-Input Line-Entry System (SMILES) uses "@" and "@@" tokens to denote chirality, while more robust representations like SELFIES and GroupSELFIES have native stereochemical tokens [23].
- Generative Models: Machine learning models, including reinforcement learning (RL) and genetic algorithms (GAs), are trained on databases like ZINC15 to generate novel molecular structures with targeted properties [23]. Stereochemistry-aware models explicitly incorporate stereochemical information during the generation process, which is crucial for optimizing stereochemistry-sensitive properties like binding affinity and optical activity [23]. Research shows these models perform on par with or surpass conventional models in such tasks, despite the increased complexity of the chemical search space [23].
Louis Pasteur's meticulous observation of tartrate crystals in 1848 unveiled the fundamental principle of molecular chirality, creating the field of stereochemistry. His discovery that the spatial arrangement of atoms is a critical determinant of molecular function has evolved from a foundational concept in organic chemistry to an indispensable pillar of modern pharmaceutical science. Today, from the design of 3D-enriched screening libraries and stereospecific SAR studies to the application of stereochemistry-aware AI generative models and stringent regulatory requirements, Pasteur's legacy endures. The journey from his crystals to contemporary drug development underscores a continuous thread: in the chiral world of biology, the correct three-dimensional structure is not a mere detail—it is often the very key to efficacy and safety.
Within the framework of stereochemistry and isomerism research, the existence of meso compounds presents a fascinating paradox that challenges initial assumptions about molecular chirality. By definition, a meso compound is an achiral molecule that possesses two or more chiral centers, yet is optically inactive and superimposable on its mirror image [38] [39]. This apparent contradiction to the fundamental rule that chiral centers impart chirality is resolved by the presence of specific symmetry elements within the molecule's structure [40]. For researchers and drug development professionals, understanding meso compounds is not merely an academic exercise; it is crucial for predicting optical activity, interpreting spectroscopic data, and designing syntheses where stereochemistry dictates biological activity. The most classic example is found in tartaric acid, which exists in two enantiomeric forms and one meso form, each with distinct physical properties [41]. This guide delves into the structural basis, identification protocols, and research implications of these exceptional molecules.
Structural Basis and Defining Symmetry Elements
The achirality of a meso compound arises from the presence of an internal plane of symmetry (also called a mirror plane) that bisects the molecule into two mirror-image halves [38] [42]. This symmetry element makes one half of the molecule the mirror reflection of the other, effectively canceling out the optical activity typically induced by the chiral centers [40].
A meso compound must fulfill three key criteria:
- It must contain two or more stereocenters (chiral centers) [38].
- It must possess an internal plane of symmetry [38] [43].
- It must contain stereocenters with identical substituents [38]. The chiral centers must have opposite configurations (one R and one S), leading to a net cancellation of optical rotation [38] [44].
A critical practice for researchers is recognizing that molecular conformation can obscure the plane of symmetry. Single bonds can rotate, and a molecule that appears asymmetric in one drawing may reveal a symmetry plane in a different, energetically accessible conformation [40] [44]. Therefore, analysis must consider the molecule's ability to adopt a conformation with a symmetry plane, not just its static representation.
Methodologies for Identification and Analysis
Visual Identification and the Role of Fischer Projections
Fischer projections provide a two-dimensional framework that greatly simplifies the identification of meso compounds [43]. The key is to look for a plane of symmetry that cuts through the molecule horizontally or vertically. For instance, in 2,3-dichlorobutane, the meso isomer is the one where the horizontal reflection of the top half perfectly reproduces the bottom half [43].
A common pitfall, often called the "Meso Trap," is assuming that two differently drawn structures are enantiomers when they are, in fact, the same meso molecule [44]. Two structures drawn as mirror images of a meso compound are superimposable, meaning they represent identical molecules, not a pair of enantiomers [44]. This is empirically verified by mentally rotating one representation 180° to see if it matches the other [38] [41].
Absolute Configuration (R/S) Analysis
A robust methodological approach to confirm a meso compound is to assign the absolute configuration (R or S) to each chiral center [44]. In a meso compound with two stereocenters, the configurations will be inverted relative to each other—specifically, one will be R and the other S [38] [44]. This (R,S) designation is a necessary but not sufficient condition; the substituents on the chiral centers must also be identical [44]. For example, while (2R,3S)-2,3-dibromobutane is meso, (2S,3R)-2-bromo-3-methylpentane is not, because the chiral centers lack identical substituents [44].
Table 1: Key Characteristics of Molecule Types with Multiple Stereocenters
| Characteristic | Meso Compound | Enantiomeric Pair | Diastereomer |
|---|---|---|---|
| Number of Stereoisomers | One distinct molecule [38] | Two mirror-image molecules [38] | Multiple, non-mirror-image molecules [45] |
| Superimposable on Mirror Image? | Yes [39] | No [45] | No |
| Internal Plane of Symmetry? | Yes [38] | No [38] | No |
| Optical Activity | Inactive [38] | Active (equal and opposite rotation) [45] | Varies |
| R/S Relationship | (R,S) or (S,R) [44] | (R,R) and (S,S) [44] | Differs at some, but not all, centers |
Experimental Characterization and Protocols
Experimental Determination of Optical Activity
The definitive experimental protocol for distinguishing a meso compound from a chiral one is polarimetry [45]. A polarimeter measures a compound's ability to rotate the plane of plane-polarized light.
Protocol:
- Preparation: Prepare a solution of the analyte of known concentration in an achiral solvent.
- Baseline: Pass plane-polarized light through a sample cell containing only the pure solvent to establish a baseline reading (α_solvent).
- Measurement: Replace the solvent with the analyte solution and measure the observed rotation (α_observed).
- Calculation: Calculate the specific rotation [α] using the formula: [α] = α_observed / (c * l), where c is concentration (g/mL) and l is path length (dm).
- Interpretation: A meso compound will yield a specific rotation of 0° (optically inactive), whereas a single enantiomer will show a non-zero value [α], and a racemic mixture will also yield 0° [45].
It is critical to note that while both meso compounds and racemic mixtures are optically inactive, they are fundamentally different. A meso compound is a single, pure achiral substance, while a racemic mixture is a 50:50 mixture of two chiral enantiomers [39] [45].
Physical Property Analysis
Beyond optical activity, meso compounds can be distinguished from their chiral diastereomers through their physical properties [41]. Since meso compounds and enantiomers are not mirror images, they are diastereomers of each other and thus have different physical properties.
Table 2: Comparative Physical Data for Tartaric Acid Stereoisomers [41]
| Stereoisomer | Specific Rotation [α] | Melting Point (°C) | Solubility (g/100 mL H₂O) |
|---|---|---|---|
| L-(+)-Tartaric Acid | +12° | 170 | 139 |
| D-(-)-Tartaric Acid | -12° | 170 | 139 |
| Meso-Tartaric Acid | 0° | 146 | 125 |
This data demonstrates that while enantiomers share identical physical properties like melting point and solubility, the meso diastereomer has distinct characteristics, making it separable by standard techniques like crystallization [45].
Essential Research Reagents and Materials
Table 3: Research Reagent Solutions for Meso Compound Analysis
| Reagent / Material | Function/Application in Research |
|---|---|
| Polarimeter | The primary instrument for determining optical activity by measuring the rotation of plane-polarized light [45]. |
| Achiral Solvents (e.g., Hexane, Acetone, Methanol) | Used to prepare samples for polarimetry and chromatography without introducing a chiral environment [45]. |
| Chiral Derivatizing Agents (e.g., MPA, MTPA) | Used to convert enantiomeric mixtures into diastereomers via chemical reaction, allowing for separation by NMR or achiral chromatography [45]. |
| Chiral Stationary Phase (HPLC/GC) | Used for the direct chromatographic separation of enantiomers, which is not required for a pure meso compound but is essential for analyzing mixtures containing meso and chiral forms [45]. |
Application in Pharmaceutical and Chemical Research
The implications of meso compounds in drug development are profound. Biological systems are chiral, and proteins, enzymes, and receptors interact differently with each enantiomer of a chiral drug [45]. A meso compound, being achiral, will not have enantiomer-specific biological activity. This can be a significant advantage, as it eliminates concerns about one enantiomer being therapeutically active while the other is inactive or toxic, a notorious example being the drug thalidomide [45].
Furthermore, the presence of a meso form affects the total number of possible stereoisomers for a given molecular structure. For a molecule with n chiral centers, the theoretical maximum number of stereoisomers is 2^n. However, if one of these is a meso compound, the actual number is 2^n - 1 [39]. This is critical for synthetic chemists planning stereoselective syntheses and calculating reaction yields, as the meso form can be a unique and predictable product, especially in symmetric reactions. For instance, hydrogenation of a symmetric diketone might produce a meso diol as a single stereoisomer, while an asymmetric counterpart would yield a pair of enantiomers requiring separation.
The thalidomide disaster represents a pivotal case study in medicinal chemistry, demonstrating the critical importance of stereochemistry in drug development and safety. This technical analysis examines the mechanistic underpinnings of thalidomide's teratogenicity through the lens of stereoisomerism, exploring how apparently subtle spatial arrangements of atoms confer dramatically different biological activities. We detail the molecular interactions between thalidomide enantiomers and their primary biological target, cereblon, and present quantitative data on the stereospecific binding affinities that underpin the drug's tragic teratogenic effects. Furthermore, we analyze the phenomenon of in vivo racemization and its implications for pharmaceutical development, providing experimental methodologies for investigating stereochemical properties of chiral therapeutics. This case continues to inform regulatory frameworks and drug design paradigms six decades after the initial tragedy, serving as an enduring lesson in molecular pharmacology.
Stereochemistry, the study of the three-dimensional arrangement of atoms in molecules, represents a fundamental domain in organic chemistry and drug development with profound implications for biological activity [46]. Chirality, formally defined as the geometric property of a rigid object of not being superimposable on its mirror image, is ubiquitous throughout biological systems [3]. Chiral molecules exist as pairs of enantiomers - non-superimposable mirror images that share identical two-dimensional structural formulas but differ in their three-dimensional orientation [47].
In achiral environments, enantiomers exhibit identical physical and chemical properties. However, in biological systems - which are inherently chiral due to the homochirality of biomolecules such as L-amino acids and D-sugars - each enantiomer may display distinct pharmacological behavior [3]. This dichotomy arises from the precise structural complementarity required for molecular recognition at biological target sites; just as a left hand fits poorly into a right-handed glove, one enantiomer may bind effectively to a protein binding site while its mirror image may not [3].
The thalidomide molecule contains a single stereogenic center, making it capable of existing as two enantiomers: (R)-thalidomide and (S)-thalidomide [48]. Initially marketed as a racemic mixture (containing equal amounts of both enantiomers), the drug was prescribed as a sedative and antiemetic for morning sickness in pregnant women before its teratogenic properties were recognized [49]. The tragic consequences of this oversight would forever change pharmaceutical regulation and underscore the critical importance of stereochemical considerations in drug development.
Historical Context of the Thalidomide Disaster
Pharmaceutical Development and Initial Use
Thalidomide was first synthesized in 1953 by Swiss pharmaceutical company CIBA and subsequently introduced by German company Chemie Grünenthal in 1956 as a non-barbiturate sedative hypnotic [49]. Marketed under the brand name Contergan, the drug was promoted as capable of producing deep sleep without hangover effects or risk of dependency [49]. Preclinical testing in rodent models failed to establish a median lethal dose, leading to the widespread belief that the drug was non-toxic to humans [49]. During this era, formal testing for teratogenic effects was not standard practice in pharmaceutical development.
Thalidomide gained rapid popularity worldwide, particularly among pregnant women for its effective anti-emetic properties in managing morning sickness [49]. The drug's accessibility without prescription and relatively low cost contributed to its widespread use, with Germany alone consuming an estimated 14.6 tons of thalidomide in 1960 [49]. In the United States, the drug was briefly available as an investigational agent but never received formal approval for marketing, largely due to the interventions of FDA officer Dr. Frances Kelsey [49].
Emergence of Teratogenicity and Global Impact
In 1961, independent observations by Australian obstetrician Dr. William McBride and German pediatrician Dr. Widukind Lenz linked thalidomide use during pregnancy to severe congenital malformations [49]. These observations were subsequently confirmed by cases reported worldwide, leading to the drug's eventual withdrawal from most commercial markets by 1961 and a worldwide ban by the end of the decade [49].
The teratogenic effects manifested primarily as limb malformations (amelia, phocomelia, syndactyly), though additional abnormalities included atresia of the esophagus, duodenum, and anus, cardiac defects, and aplasia of the gallbladder and appendix [49]. The critical period for teratogenic susceptibility occurred between 34 and 49 days after the last menstrual period, with even single doses associated with significantly increased risk [49]. Up to 40% of affected infants died within their first year of life [49].
The global impact was staggering: an estimated 10,000 infants were affected worldwide, with additional uncounted stillbirths and miscarriages [49]. The United States was largely spared from the catastrophe due to Dr. Kelsey's refusal to approve the drug based on emerging data linking thalidomide to neurological toxicities, including peripheral neuritis [50]. For her vigilance, Dr. Kelsey received the President's Award for Distinguished Federal Civilian Service from President John F. Kennedy in 1962 [49].
Table 1: Timeline of Key Events in the Thalidomide Disaster
| Year | Event |
|---|---|
| 1953 | Thalidomide first synthesized by CIBA [49] |
| 1956 | Introduced by Chemie Grünenthal as Contergan [49] |
| 1957-1961 | Widespread use as sedative and antiemetic [49] |
| 1961 | McBride and Lenz independently identify teratogenic link [49] |
| 1961 | Withdrawn from most markets [49] |
| 1962 | Dr. Frances Kelsey receives presidential award [49] |
| 1960s | Banned worldwide [49] |
Stereochemical Properties of Thalidomide
Molecular Structure and Enantiomeric Characteristics
Thalidomide, α-(N-phthalimido) glutarimide, is a racemic derivative of glutamic acid consisting of equal amounts of the (R)-(+) and (S)-(-) enantiomers [49]. The compound features a single stereogenic center at the glutarimide ring, granting it chirality and the capacity to exist as two enantiomeric forms [51]. The (R)-enantiomer was initially associated with sedative effects, while the (S)-enantiomer was identified as teratogenic [48].
A critical aspect of thalidomide's stereochemistry is the rapid chiral interconversion that occurs under physiological conditions [49]. The enantiomers undergo spontaneous hydrolysis and interconversion in vivo, with the half-life for racemization measured at approximately 2.5 hours in phosphate buffer and even faster (approximately 1 hour) in serum [52]. This dynamic interconversion means that administering either pure enantiomer ultimately results in exposure to both forms within biological systems.
Table 2: Properties of Thalidomide Enantiomers
| Property | (R)-Thalidomide | (S)-Thalidomide |
|---|---|---|
| Optical Rotation | (+) dextrorotatory [51] | (-) levorotatory [51] |
| Primary Pharmacological Effect | Sedative [48] | Teratogenic [48] |
| TNF-α Inhibition | Minimal effect [49] | Potent inhibitor [49] |
| Cereblon Binding Affinity | Lower affinity [52] | 10-fold stronger binding [52] |
| Anti-angiogenic Activity | Present but weaker [49] | More potent [49] |
The "Thalidomide Paradox" and Self-Disproportionation
The phenomenon known as the "thalidomide paradox" refers to the apparent contradiction between the observed differences in biological activity of the individual enantiomers and their rapid in vivo interconversion [52]. If the enantiomers readily interconvert under physiological conditions, then animal experiments using pure (R)-thalidomide should theoretically result in teratogenicity due to racemization, yet such studies demonstrated that (R)-thalidomide alone did not produce teratogenic effects [52].
Recent research has proposed a resolution to this paradox through the phenomenon of self-disproportionation of enantiomers (SDE). Experiments have demonstrated that when non-racemic thalidomide mixtures are suspended in aqueous solutions (including biological media), significant enantiomeric enrichment occurs in the supernatant while racemic thalidomide precipitates in (R/S)-heterodimeric form [52]. In one experimental setup, an initial 19% enantiomeric excess (ee) of (R)-thalidomide resulted in solutions with up to 98% ee after stirring in phosphate buffer [52]. This selective precipitation effectively removes racemic thalidomide from biological availability while leaving enantiomerically enriched material in solution, potentially explaining why administration of pure (R)-thalidomide does not necessarily lead to teratogenic effects despite in vivo racemization.
Mechanisms of Teratogenic Action
Cereblon Binding and SALL4 Degradation
The molecular mechanism underlying thalidomide's teratogenicity remained elusive for decades until landmark studies identified cereblon (CRBN) as the primary molecular target [53]. Cereblon functions as a substrate receptor for the CUL4-RBX1-DDB1-CRBN E3 ubiquitin ligase complex, which mediates the ubiquitination and subsequent proteasomal degradation of specific protein substrates [53].
Structural and biochemical studies have revealed that thalidomide binds to a hydrophobic pocket within the cereblon protein, with the (S)-enantiomer exhibiting approximately 10-fold stronger binding affinity and more effective inhibition of cereblon's self-ubiquitylation compared to the (R)-isomer [52]. This binding event alters the substrate specificity of the E3 ubiquitin ligase complex, leading to the recruitment and degradation of developmentally critical transcription factors that would not normally be targeted [53].
A key teratogenic mechanism involves the cereblon-mediated degradation of the transcription factor SALL4 [53]. Degradation of SALL4 interferes with limb development and other aspects of fetal growth, resulting in the spectrum of complications indelibly linked to thalidomide: deformed limbs, defective organs, and the other characteristic malformations [53]. This mechanism is strongly supported by clinical observations that individuals with mutations in the SALL4 gene exhibit congenital abnormalities strikingly similar to those seen in thalidomide-exposed children, including missing thumbs, underdeveloped limbs, eye and ear defects, and congenital heart disease [53].
Diagram 1: Teratogenic pathway of SALL4 degradation
Anti-angiogenic and Immunomodulatory Effects
In addition to cereblon-mediated mechanisms, thalidomide exhibits potent anti-angiogenic properties that contribute to its teratogenic effects [49]. The anti-angiogenic activity was first demonstrated using a rabbit cornea model of basic fibroblast growth factor (bFGF)-induced neovascularization [49]. Researchers postulated that thalidomide-induced birth defects resulted from inhibited blood vessel growth in the developing fetal limb bud, with similar mechanisms potentially inhibiting vasculogenesis in the tumor microenvironment [49].
The immunomodulatory properties of thalidomide also play significant roles in its biological activity. One primary effect is the selective inhibition of TNF-α production in human monocytes, achieved through enhanced degradation of TNF-α mRNA [49]. Additionally, thalidomide inhibits nuclear factor kappa-B (NF-κB) activity through a process involving inhibition of I-κB kinase activity [49]. NF-κB is a DNA binding transcription factor that regulates expression of genes contributing to immune responses, including TNF-α, interleukin (IL)-8, and IL-12 [49].
Thalidomide also modulates T-cell responses by stimulating T-cell proliferation with preferential expansion in the CD8+ T-cell subset and shifting T-helper cell responses from a Th1 to Th2 cytokine profile [49]. This immunomodulation involves enhanced IL-4 production and inhibited interferon (IFN)-γ production in peripheral mononuclear cells [49].
Table 3: Key Mechanisms of Thalidomide Action
| Mechanism | Molecular Target/Effect | Biological Consequence |
|---|---|---|
| Cereblon Binding | Alters E3 ubiquitin ligase specificity [53] | Degradation of SALL4 and other transcription factors [53] |
| Anti-angiogenesis | Inhibits bFGF/VEGF-induced neovascularization [49] | Impaired limb bud development [49] |
| TNF-α Inhibition | Enhances TNF-α mRNA degradation [49] | Reduced inflammation [49] |
| NF-κB Inhibition | Suppresses I-κB kinase activity [49] | Decreased pro-inflammatory cytokine production [49] |
| T-cell Modulation | Shifts Th1 to Th2 cytokine profile [49] | Enhanced IL-4, reduced IFN-γ production [49] |
Experimental Approaches and Analytical Methods
Chromatographic Enantioseparation Protocols
The analysis of thalidomide enantiomers requires specialized chromatographic methods capable of chiral separation. High-performance liquid chromatography (HPLC) using chiral stationary phases represents the most reliable approach for determining enantiomeric purity and studying racemization kinetics.
Standard Protocol for Chiral HPLC Analysis:
- Column: Chiralpak AD, AS, or equivalent chiral stationary phase
- Mobile Phase: n-Hexane/Isopropanol/Trifluoroacetic Acid (70:30:0.1 v/v/v)
- Flow Rate: 1.0 mL/min
- Detection: UV at 220 nm
- Temperature: 25°C
- Injection Volume: 10 μL
This method typically achieves baseline separation of (R)- and (S)-thalidomide with resolution factors >1.5, enabling accurate quantification of enantiomeric composition in pharmaceutical formulations and biological samples [52].
Racemization Kinetics Studies
Understanding the interconversion kinetics between thalidomide enantiomers is essential for predicting biological behavior. The following protocol characterizes racemization under physiological conditions:
Racemization Kinetics Protocol:
- Prepare separate solutions of pure (R)- or (S)-thalidomide in phosphate-buffered saline (PBS, pH 7.4) at 37°C.
- Withdraw aliquots at predetermined time intervals (0, 0.5, 1, 2, 4, 8, 12, 24 hours).
- Immediately analyze aliquots by chiral HPLC to determine enantiomeric ratio.
- Plot enantiomeric excess versus time to determine racemization half-life.
Using this approach, researchers have determined that thalidomide undergoes rapid chiral interconversion with a half-life of approximately 1-2 hours in serum and 12 hours in buffer solutions at physiological pH [52].
Self-Disproportionation of Enantiomers (SDE) Experiments
The investigation of SDE phenomena provides insights into the "thalidomide paradox" and involves the following methodology:
SDE Experimental Procedure:
- Prepare non-racemic mixtures of thalidomide enantiomers (e.g., 20% enantiomeric excess).
- Suspend the solid mixture in aqueous media (water or phosphate buffer, pH 7.4).
- Stir vigorously for predetermined periods (1-24 hours) at physiological temperature (37°C).
- Separate supernatant from precipitate by filtration or centrifugation.
- Determine enantiomeric composition of both fractions by chiral HPLC.
- Compare results to initial composition to quantify enrichment factors.
This methodology has demonstrated that significant enantiomeric enrichment occurs in solution, with ee values increasing from 20% to over 90% in some cases, while the precipitate exhibits near-racemic composition [52].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagents for Thalidomide Stereochemistry Studies
| Reagent/Material | Specifications | Research Application |
|---|---|---|
| Chiral HPLC Columns | Chiralpak AD, AS; 250 × 4.6 mm; 5 μm particle size | Enantioseparation and purity assessment [52] |
| Deuterated Enantiomers | Deuterium-substituted at chiral center | Studying enantiomer-specific effects without interference from racemization [52] |
| Recombinant Cereblon Protein | CRBN-binding domain, >95% purity | Binding affinity studies and crystallography [52] |
| SALL4 Antibodies | Monoclonal, validated for Western blot | Detection of SALL4 degradation in cell-based assays [53] |
| Chiral Solvents | HPLC grade n-hexane, isopropanol | Mobile phase preparation for chiral separations [52] |
| Ultrafiltration Devices | 10 kDa molecular weight cut-off | Protein binding studies and free drug concentration determination |
Contemporary Relevance and Regulatory Impact
Modern Therapeutic Applications
Despite its tragic history, thalidomide has experienced a remarkable therapeutic renaissance, earning FDA approval for at least 13 different indications [51]. The drug's immunomodulatory and anti-angiogenic properties have proven valuable in treating conditions including erythema nodosum leprosum, multiple myeloma, graft-versus-host disease, Behçet's syndrome, and Crohn's disease [51]. This resurgence, however, necessitates strict risk evaluation and mitigation strategies (REMS) to prevent fetal exposure, particularly the Thalidomide REMS program which mandates contraception requirements and regular pregnancy testing for females of reproductive potential.
The understanding of thalidomide's mechanism of action has spurred development of analogues with improved safety profiles, such as lenalidomide and pomalidomide [49]. These derivatives maintain therapeutic efficacy while demonstrating reduced teratogenic potential, though they remain contraindicated in pregnancy due to shared mechanistic pathways with the parent compound.
Legacy in Pharmaceutical Regulation
The thalidomide disaster precipitated fundamental changes in drug regulatory frameworks worldwide. The 1962 Kefauver-Harris Amendments to the United States Federal Food, Drug, and Cosmetic Act mandated that drug manufacturers prove efficacy in addition to safety before receiving marketing approval [50]. These amendments also required:
- Informed consent from clinical trial participants
- Adverse event reporting requirements
- Good Manufacturing Practice regulations
- FDA authority to withdraw approval for drugs not demonstrating substantial evidence of efficacy
The catastrophe also highlighted limitations in animal models for predicting human teratogenicity, stimulating research into more predictive toxicological approaches and the development of guidelines for reproductive toxicity testing [49].
Diagram 2: Regulatory evolution after thalidomide
The thalidomide disaster stands as a seminal lesson in pharmaceutical development, permanently underscoring the critical importance of stereochemical considerations in drug design, testing, and regulation. The complex stereodynamic behavior of thalidomide - including its rapid in vivo racemization and the self-disproportionation of its enantiomers - illustrates that chiral drugs must be evaluated as dynamic three-dimensional systems rather than static structural diagrams. The resolution of the "thalidomide paradox" through contemporary research methodologies demonstrates how persistent scientific inquiry can unravel even the most challenging pharmacological mysteries decades after their emergence.
From a regulatory perspective, the tragedy catalyzed essential reforms that continue to protect public health today, establishing rigorous standards for drug safety and efficacy demonstration. For medicinal chemists and pharmaceutical researchers, thalidomide remains a cautionary tale that informs screening approaches, analytical methodologies, and formulation strategies for chiral therapeutics. As drug development increasingly focuses on targeted therapies and sophisticated molecular design, the stereochemical lessons from thalidomide maintain their relevance, ensuring that the profound suffering caused by this molecule ultimately contributes to safer, more effective pharmaceutical interventions.
Analytical Techniques and Pharmaceutical Applications of Stereochemistry
The Cahn-Ingold-Prelog (CIP) priority rules constitute a fundamental systematic approach for unambiguously describing the three-dimensional arrangement of atoms around stereogenic elements in organic molecules. This in-depth technical guide examines the CIP system's theoretical foundations, detailed procedural protocols for assigning absolute configurations, and its critical applications in pharmaceutical development and environmental chemistry. By providing standardized methodology for designating stereochemistry at chiral centers (R/S) and double bonds (E/Z), the CIP framework enables precise structural communication essential for understanding structure-activity relationships in drug molecules, predicting physicochemical properties of stereoisomers, and ensuring regulatory compliance in the development of chiral therapeutics. This review integrates the official IUPAC rules with practical implementation strategies, experimental protocols for configuration assignment, and contemporary research applications that underscore the system's enduring significance in modern stereochemical analysis.
Stereochemistry represents a cornerstone of organic chemistry that investigates the spatial arrangement of atoms and their profound influence on molecular properties and reactivity. Within this domain, isomerism—the phenomenon where molecules share identical atomic composition but differ in structure—manifests in several distinct forms. Constitutional isomers possess the same molecular formula but differ in atomic connectivity, while stereoisomers share identical atomic connectivity but differ in the three-dimensional orientation of their atoms in space [54].
Stereoisomers are further categorized into two primary classes:
- Enantiomers: Non-superimposable mirror images of each other, typically characterized by chiral centers where a carbon atom bears four different substituents [55] [54]
- Diastereomers: Stereoisomers that are not mirror images, including geometric isomers arising from restricted rotation around double bonds or rings [54]
The biological activity of organic molecules, particularly pharmaceuticals, frequently depends on their stereochemical configuration. Chiral recognition in biological systems means that enantiomers can exhibit dramatically different pharmacological profiles, metabolic pathways, and toxicological effects [56]. This fundamental principle underscores the critical importance of unambiguous stereochemical description systems in chemical research and development.
Theoretical Framework of the CIP System
Historical Development and Conceptual Foundations
The CIP system was developed in the mid-20th century by organic chemists Robert Sidney Cahn, Christopher Kelk Ingold, and Vladimir Prelog to address the pressing need for a universal, unambiguous method to specify absolute molecular configurations [57] [56]. Prior to its introduction, stereochemical description relied on relative notations like the D/L system, which was limited to specific compound classes such as sugars and amino acids [56].
The CIP convention was formally adopted by the International Union of Pure and Applied Chemistry (IUPAC) in 1974 as the standard for stereochemical nomenclature [57]. The system has undergone subsequent revisions, most recently in 2013, to address complex molecular scenarios and maintain its applicability across the expanding frontier of chemical space [57]. The framework operates through a hierarchical set of sequence rules that assign priority to substituents based on atomic properties, enabling the precise specification of configuration at stereocenters and double bonds.
The CIP Priority Rules: Core Principles
The CIP system employs a deterministic algorithm for ranking substituents attached to stereogenic elements. The foundational rules proceed sequentially, with each subsequent rule applied only when prior rules cannot break ties:
Rule 1: Atomic Number Priority - Compare the atomic number (Z) of atoms directly bonded to the stereocenter. The atom with the higher atomic number receives higher priority. Hydrogen (Z=1) consistently assumes the lowest priority in organic molecules [57] [58] [59].
Rule 2: Isotopic Mass Priority - For identical atoms differing only in isotopic composition, the isotope with higher mass receives higher priority (e.g., deuterium > hydrogen, ¹³C > ¹²C) [57] [60].
Rule 3: Chain Extension for Tie-Breaking - When two substituents have identical first atoms, compare subsequent atoms along the chains. Lists of attached atoms are compiled in order of decreasing atomic number and compared atom-by-atom at the first point of difference [57] [55].
Rule 4: Multiple Bond Handling - Double and triple bonds are expanded by treating them as if the atom is bonded to multiple phantom atoms of the bonded partner. For example, a carbonyl carbon (C=O) is treated as being bonded to two oxygen atoms, while a nitrile carbon (C≡N) is treated as being bonded to three nitrogen atoms [57] [59] [61].
Table 1: Atomic Number Priority of Common Elements in Organic Molecules
| Element | Atomic Number (Z) | Priority Rank | Example Functional Groups |
|---|---|---|---|
| Iodine | 53 | 1 | Iodomethane, alkyl iodides |
| Bromine | 35 | 2 | Bromoethane, alkyl bromides |
| Chlorine | 17 | 3 | Chloromethane, alkyl chlorides |
| Oxygen | 8 | 4 | Alcohols, ethers, carbonyls |
| Nitrogen | 7 | 5 | Amines, amides, nitriles |
| Carbon | 6 | 6 | Alkyl chains, aromatics |
| Hydrogen | 1 | 7 | C-H bonds |
The rule application process continues recursively outward from the stereocenter until all ties are broken. For cyclic molecules, the system employs a hierarchical digraph approach that traverses all possible paths from the stereocenter, generating phantom atoms when paths encounter previously visited atoms to maintain a finite tree structure [57].
Experimental Protocols for Configuration Assignment
Step-by-Step Methodology for R/S Determination
The assignment of absolute configuration to a chiral center follows a systematic procedure that integrates the CIP priority rules with spatial analysis:
Identify the Stereocenter: Locate the tetrahedral carbon atom with four different substituents [55].
Assign Priorities: Apply the CIP rules to rank the four substituents from highest (1) to lowest (4) priority [58] [59].
Orient the Molecule: Position the molecule such that the lowest priority group (4) points away from the observer. In standard wedge-dash notation, this corresponds to the substituent on a dashed bond [55] [58].
Trace the Sequence: Draw a curved arrow from priority 1 → 2 → 3, ignoring the fourth substituent [55] [59].
Assign Configuration:
Diagram 1: CIP Configuration Assignment Workflow
Special Cases and Troubleshooting Protocols
Handling Non-Ideal Molecular Orientations: When the lowest priority group is not conveniently positioned on a dashed bond, employ these experimental workarounds:
- #4 on Wedge: Apply the reverse rule—clockwise = S, counterclockwise = R [55] [61]
- #4 in Plane: Use the "single swap" method—swap #4 with the back substituent, determine configuration on the modified structure, then reverse the result (R⇔S) [55] [61]
Breaking Complex Ties with the Dot Technique: For substituents with identical first atoms, mark each atom with a "dot" and list subsequent attached atoms in descending order of atomic number. Compare these lists lexicographically at the first point of difference [55].
Table 2: Troubleshooting Common CIP Assignment Challenges
| Scenario | Challenge | Recommended Protocol | Example Application |
|---|---|---|---|
| Multiple carbon substituents | Tie-breaking at first atom | Dot technique with atomic number comparison | -CH3 (H,H,H) vs -CH2OH (O,H,H) |
| Double/triple bonds | Proper priority assignment | Multiple bond expansion to phantom atoms | Aldehyde carbon treated as C(O,O,H) |
| Isotopic labeling | Distinguishing isotopes | Higher mass receives priority | Deuterium > hydrogen |
| #4 group in plane | Difficult spatial visualization | Single swap method with inversion | Switch two groups, assign, then reverse |
| Complex cycles | Infinite path concerns | Hierarchical digraph with phantom atoms | Cyclic molecule traversal |
E/Z Assignment for Double Bonds
The CIP system extends to alkene stereochemistry through a related protocol:
- Analyze Each Carbon Separately: Apply CIP rules independently to the two substituents on each double-bonded carbon [57] [56]
- Identify Highest Priority Groups: Determine which substituent on each carbon has higher priority [57]
- Assign Configuration:
This system supersedes the older cis/trans notation, which fails for complex substituents where no reference hydrogen exists [54] [56].
Pharmaceutical and Research Applications
Drug Development and Stereochemical Specificity
The CIP system provides the foundational language for describing stereochemistry in pharmaceutical compounds, where configurational differences frequently dictate therapeutic efficacy and safety profiles. Notable examples include:
- Thalidomide: The (R)-enantiomer possesses sedative properties, while the (S)-enantiomer is teratogenic [56]
- Ibuprofen: The (S)-(+)-enantiomer delivers the primary anti-inflammatory activity, though the drug is typically administered as a racemate due to in vivo interconversion [56]
- Tamoxifen: The E-isomer functions as a selective estrogen receptor modulator for breast cancer treatment, while the Z-isomer is therapeutically inactive [56]
Regulatory agencies including the FDA require explicit stereochemical description for chiral therapeutics, mandating comprehensive characterization of each enantiomer's pharmacological and toxicological profile [56]. The CIP system enables this precise communication in drug applications, manufacturing documentation, and clinical literature.
Environmental Chemistry and Atmospheric Sciences
Stereochemical considerations extend to environmental processes, where isoprene oxidation in the atmosphere generates secondary organic aerosols (SOA) with potential stereochemical preferences [62]. The formation of 2-methyltetrols through atmospheric isoprene epoxidation demonstrates how stereochemical outcomes may influence aerosol properties including hygroscopicity and climate effects [62]. Chamber studies investigating SOA formation under controlled conditions rely on CIP descriptors to characterize the stereochemistry of oxidation products and their environmental fate [62].
Research Reagent Solutions and Experimental Tools
Table 3: Essential Research Materials for Stereochemical Analysis
| Reagent/Equipment | Function in Stereochemical Research | Application Example |
|---|---|---|
| Chiral stationary phase HPLC columns | Enantiomer separation and analysis | Determination of enantiomeric excess |
| Polarimeter | Measurement of optical rotation | Experimental correlation with absolute configuration |
| X-ray crystallography equipment | Direct determination of absolute configuration | Reference standard establishment |
| Molecular modeling software | 3D visualization and priority assignment | CIP practice and education |
| Physical molecular model kits | Spatial relationship visualization | Training in R/S assignment |
| Isotopically labeled compounds | Isotopic priority applications | Mechanistic tracer studies |
| Chiral resolving agents | Diastereomer formation for separation | Enantiopure material preparation |
The Cahn-Ingold-Prelog priority rules represent an indispensable framework in stereochemical analysis, providing researchers with a systematic, unambiguous method for describing molecular configuration. Its rigorous hierarchical approach enables consistent communication of structural information across chemical disciplines, from pharmaceutical development to environmental chemistry. As research continues to reveal the profound implications of stereochemistry in biological systems, materials science, and environmental processes, the CIP system remains foundational for precise stereochemical description. Future advancements may further refine the rules to address increasingly complex molecular architectures, yet the core principles established by Cahn, Ingold, and Prelog will continue to underpin stereochemical discourse and discovery.
The determination of absolute configuration (AC) and conformational properties is a fundamental challenge in the structural characterization of chiral molecules, particularly in pharmaceutical and natural product research. Chiroptical spectroscopy, which measures the differential interaction of chiral compounds with circularly polarized light, has emerged as a powerful suite of tools for addressing this challenge [63] [3]. Within this domain, Electronic Circular Dichroism (ECD) and Vibrational Circular Dichroism (VCD) have become cornerstone techniques. Their importance is magnified by the established fact that enantiomers can exhibit drastically different biological activities, pharmacological effects, and metabolic pathways [3] [64]. For instance, in pharmaceuticals, one enantiomer may be therapeutically active while its mirror image could be inactive or even harmful [3]. This technical guide outlines the core principles, practical methodologies, and advanced applications of ECD and VCD spectroscopy, providing a contemporary resource for researchers navigating the complexities of stereochemistry in organic molecules.
Fundamental Principles and Complementary Information
ECD and VCD, while both being chiroptical techniques, probe different molecular phenomena and provide distinct, yet often complementary, information about a molecule's stereochemistry.
Electronic Circular Dichroism (ECD) measures the difference in absorption of left- and right-circularly polarized light by a chiral molecule during electronic transitions [64] [65]. ECD is a sensitive technique that requires only small amounts of sample (typically ~1 mg or less) and is exceptionally fast in terms of measurement time [64]. However, the information it provides is inherently linked to the molecule's chromophore and its immediate surroundings. This means ECD is highly sensitive to the electronic environment but may not report directly on the chirality of the entire molecular skeleton, especially for UV-transparent compounds [63] [64].
Vibrational Circular Dichroism (VCD), in contrast, probes the differential absorption for vibrational transitions within the electronic ground state [64]. Its key advantage lies in the wealth of structural information it provides; since functional groups responsible for vibrations are distributed across the entire molecule, VCD spectra contain stereochemical information about the whole molecular framework [66] [64]. While VCD measurements typically require more sample (5-10 mg) and longer collection times compared to ECD, the resulting spectra feature numerous well-resolved vibrational bands, offering a highly detailed fingerprint of molecular chirality [64].
The complementary nature of these two methods is a significant strength. ECD reports on the chiral electronic structure, often of a specific chromophoric region, whereas VCD reports on the chiral vibrational structure of the entire molecule. Their combined application substantially increases the confidence level in stereochemical assignments [64].
Table 1: Core Comparison of ECD and VCD Techniques
| Feature | Electronic Circular Dichroism (ECD) | Vibrational Circular Dichroism (VCD) |
|---|---|---|
| Physical Basis | Differential absorption during electronic transitions | Differential absorption during vibrational transitions |
| Information Content | Sensitive to chromophore and its local chiral environment | Reports on chirality of the entire molecular skeleton |
| Typical Sample Need | ~1 mg or less | 5-10 mg |
| Measurement Speed | Fast (minutes) | Slower (can be hours) |
| Key Strength | High sensitivity; empirical/chromophore-based rules | High informational density; direct non-empirical assignment |
| Primary Limitation | Restricted to chromophore-bearing molecules | Requires more sample and computational power |
Experimental Protocols and Methodologies
Standard Protocol for ECD Analysis
A typical ECD experiment for absolute configuration assignment follows a well-established workflow that integrates both measurement and theoretical computation.
Sample Preparation:
- Prepare a solution of the chiral compound in a suitable UV-transparent solvent (e.g., acetonitrile, water) [63].
- The concentration should be adjusted to achieve an absorbance within the optimal range for the instrument, typically ensuring that the absorbance maximum in the region of interest is between 0.5 and 1 [65].
- Use high-purity, spectroscopic-grade solvents to avoid interfering absorbance artifacts.
Data Acquisition:
- Record the ECD spectrum at room temperature using a spectropolarimeter, such as a Jasco J-815 [63].
- Purge the instrument with nitrogen gas during measurement to protect optics and reduce ozone generation [63].
- Utilize a cell with an appropriate path length (e.g., 0.1 mm for high-concentration samples) [63].
- Acquire spectra over a sufficient wavelength range (e.g., 200-400 nm) with adequate data pitch and scanning speed to resolve key Cotton effects.
Data Analysis and Interpretation:
- Process the raw spectrum by subtracting the baseline (solvent and cell).
- Analyze the signs and magnitudes of the observed Cotton effects.
- For absolute configuration assignment, compare the experimental spectrum with quantum-chemically computed spectra for the possible stereoisomers. This involves: a. Conducting a conformational search for each candidate structure. b. Optimizing the geometries of low-energy conformers using Density Functional Theory (DFT). c. Calculating the theoretical ECD spectrum for each conformer. d. Generating a Boltzmann-weighted average spectrum and comparing it to the experimental result [63].
Standard Protocol for VCD Analysis
The VCD protocol shares similarities with the ECD workflow but focuses on vibrational transitions, requiring IR-transparent solvents and more extensive computational resources.
Sample Preparation:
- Dissolve the compound in a suitable solvent (e.g., CDCl₃, DMSO-d₆) that is transparent in the mid-IR region of interest (typically 1800-900 cm⁻¹) [67].
- A higher concentration is generally required compared to ECD, often in the range of 0.1 M, depending on the path length [64].
- Use cells with path lengths designed for IR transmission, such as demountable cells with BaF₂ windows and a path length of 50-100 µm.
Data Acquisition:
- Acquire VCD and corresponding IR absorption spectra simultaneously on a dedicated VCD spectrometer.
- Collection times are significantly longer than for ECD, often ranging from 4 to 12 hours, to achieve an acceptable signal-to-noise ratio for the inherently weak VCD signal [66] [64].
- The instrument should be purged with dry, CO₂-free air to minimize interference from atmospheric water vapor and CO₂.
Data Analysis and Interpretation:
- Process the raw interferograms by Fourier transformation and subtract the solvent background.
- The analysis is heavily reliant on theoretical simulation. The standard practice involves: a. Performing a thorough conformational search. b. Optimizing all low-energy conformers using DFT (common functionals include B3PW91 and others, with basis sets like 6-31G(d)) [66]. c. Calculating the IR and VCD spectra for each optimized conformer. d. Generating the Boltzmann-averaged spectrum and comparing it directly to the experiment [66] [64].
- A good match between the experimental and calculated VCD spectrum, particularly in the sign pattern of multiple bands, allows for a definitive assignment of the absolute configuration.
Diagram 1: Complementary workflows for ECD and VCD analysis. Both pathways involve experimental measurement coupled with theoretical computation for definitive absolute configuration assignment.
Advanced Applications and Current Research Trends
Overcoming Limitations with Chiroptical Probes
A powerful approach for UV-transparent or highly flexible compounds, which are challenging for standard computational protocols, involves the use of chiroptical probes. These are achiral chromophoric units that, when covalently linked to the chiral substrate of unknown configuration, give rise to diagnostic ECD signals [63]. A prominent example is the use of 2,2′-bridged biphenyl probes, which can be attached to diols, carboxylic acids, or primary amines. Upon derivatization, the probe adopts a chiral conformation induced by the substrate, producing a distinct ECD spectrum that reveals the substrate's absolute configuration [63]. This method is particularly valuable for conformationally mobile and ECD-silent compounds, as it transforms the problem into the analysis of a rigid, chromophore-containing derivative [63].
VCD Enhancement via Low-Lying Electronic States
While VCD signals are typically weak, significant research efforts are focused on signal enhancement. A notable phenomenon is the enhancement of VCD intensity through coupling with low-lying electronic states (LLESs), particularly in chiral transition metal complexes [67]. For example, coordination of a chiral ligand to a metal ion like Co(II) can drastically amplify the VCD signals associated with the ligand's chiral environment [67]. This enhancement is attributed to the involvement of electronic transitions in the vibrational optical activity, a effect that current DFT methods are still striving to fully replicate [67]. This area represents the cutting edge of fundamental research in chiroptical spectroscopy.
The Emerging Role of Machine Learning
A transformative trend in the field is the integration of Machine Learning (ML) to address the computational bottleneck of VCD analysis. Recent studies demonstrate that ML models can be trained to predict the VCD spectrum of a molecular conformer directly from its geometry, bypassing the need for a quantum chemical calculation for every single conformer [66]. Although these models are not yet transferable between different molecular scaffolds, they offer the potential for dramatic reductions in the computational time and resources required to generate theoretical spectra, thereby accelerating the overall assignment process [66].
Table 2: Key Reagents and Materials for Chiroptical Experiments
| Reagent/Material | Function | Example Application |
|---|---|---|
| Biphenyl Dioxolane Probe | Chiroptical derivatizing agent for diols | Absolute configuration assignment of erythro and threo diols via ECD [63] |
| Dimolybdenum Tetracarboxylate | Auxiliary chromophore for vic-diols | In situ complex formation for ECD-based AC assignment (Dimolybdenum method) [64] |
| Chiral Stationary Phases (CSPs) | Enantioseparation for sample purification | Polysaccharide-based phases for HPLC purification prior to analysis [68] |
| Deuterated Solvents (CDCl₃, DMSO-d₆) | Solvent for NMR and VCD spectroscopy | Provides an IR-transparent window for VCD measurement [67] |
| Schiff Base Ligands | Chiral ligands for metal complexes | Study of chirality and VCD enhancement in transition metal complexes (e.g., Co(II)-salen) [67] |
ECD and VCD spectroscopy have matured into indispensable tools for the stereochemical characterization of chiral organic molecules. The synergy between experimental measurement and theoretical calculation provides a robust, non-empirical framework for determining absolute configuration and elucidating conformational landscapes. As the field advances, the development of chiroptical probes for challenging substrates, the exploration of enhancement mechanisms in metal complexes, and the integration of machine learning for computational efficiency are pushing the boundaries of what is possible. For researchers in drug development and natural product chemistry, mastering these advanced chiroptical methods is no longer a specialty but a core competency essential for ensuring the efficacy and safety of chiral molecules.
Stereochemistry, the study of the three-dimensional arrangement of atoms in molecules, is a fundamental concept in organic chemistry and life sciences. Chirality, a manifestation of stereochemistry, describes molecules that are non-superimposable on their mirror images, much like a left and right hand [69]. These mirror-image forms are called enantiomers. The profound significance of chirality stems from the fact that major biological activities, including molecular recognition, enzyme-substrate interactions, and receptor binding, occur through strict chiral matching [70]. Consequently, the biological activity, pharmacokinetics, and metabolic fate of a chiral molecule can differ dramatically between its enantiomers [69].
The high stakes of enantioselectivity are starkly illustrated by historical tragedies and therapeutic successes. For instance, in the case of thalidomide, one enantiomer provided the desired sedative effect, while the other was responsible for teratogenic effects [69]. Similarly, (R)-methadone acts as an analgesic, whereas its enantiomer, (S)-methadone, can bind to the hERG protein and cause severe cardiac side effects [23]. Conversely, the development of single-enantiomer drugs, such as levocetirizine (derived from cetirizine) and dexibuprofen (the active enantiomer of ibuprofen), has led to therapies with improved efficacy and reduced side-effect profiles [69]. These examples underscore that enantiomeric purity is not merely an academic pursuit but a critical determinant of safety and efficacy in pharmaceuticals, driving the continuous development of robust and efficient chiral separation technologies.
Core Principles of Chiral Separation in Chromatography
The fundamental challenge in separating enantiomers is that they possess identical physicochemical properties in an achiral environment. Their separation by chromatography, therefore, requires the transient creation of a diastereomeric relationship between the enantiomers and a chiral environment. This is most commonly achieved by using a chiral stationary phase (CSP). The CSP contains a chiral selector that interacts differentially with the two enantiomers of a analyte. The three-point interaction model is often used to explain this, proposing that for successful enantioseparation, a minimum of three simultaneous interactions must occur between the analyte and the chiral selector, with at least one of these interactions being stereochemically dependent.
The differential affinity causes one enantiomer to be retained longer on the column than the other, leading to their separation. The degree of separation is quantitatively described by two key parameters: the separation factor (α), which is the ratio of the retention factors of the two enantiomers and indicates selectivity, and the resolution (Rs), a combined measure of the efficiency and selectivity of the separation [71]. Modern chiral separation leverages advanced CSPs and sophisticated chromatographic techniques, primarily High-Performance Liquid Chromatography (HPLC) and Supercritical Fluid Chromatography (SFC), to achieve this discrimination.
High-Performance Liquid Chromatography (HPLC) for Chiral Separation
HPLC is the most widely used method for the separation and determination of enantiomers [70]. In an HPLC system, a high-pressure infusion pump drives the liquid mobile phase (which can be a single solvent or a mixture) through a column packed with the stationary phase. The sample components are separated based on their differential distribution between the mobile and stationary phases. The separated analytes then pass through a detector (e.g., Ultraviolet, Mass Spectrometry, or Charged Aerosol Detector) for identification and quantification [70] [72]. HPLC is particularly suitable for analyzing chiral materials with strong polarity and poor thermal stability [70].
Chiral Stationary Phases for HPLC
The heart of chiral HPLC is the CSP. A diverse array of chiral selectors is commercially available, each with unique recognition mechanisms suited to different classes of chiral compounds.
Table 1: Common Types of Chiral Stationary Phases for HPLC
| CSP Type | Chiral Selector Examples | Typical Recognition Mechanisms |
|---|---|---|
| Polysaccharide-Based | Coated or immobilized amylose (e.g., Chiralpak AD/IA) and cellulose (e.g., Chiralcel OD/OJ) derivatives [71] | Hydrogen bonding, dipole-dipole interactions, π-π interactions, and inclusion complexation within the helical polymer structure. |
| Macrocyclic Glycopeptides | Vancomycin, Teicoplanin, Teicoplanin Aglycone [73] [72] | Multiple interactions including ionic, hydrogen bonding, π-π, and dipole-dipole, due to their "basket-shaped" cavity. |
| Cyclodextrins | α-, β-, γ-Cyclodextrins and their derivatives (e.g., HP-β-CD) [74] [73] | Formation of inclusion complexes within the hydrophobic cavity, with enantioselectivity governed by interactions at the mouth of the cavity. |
| Pirkle (Brush-Type) | Donor-acceptor phases derived from small chiral molecules like π-acceptors or π-donors [74] [71] | Targeted, complementary interactions such as π-π interactions, hydrogen bonding, and dipole stacking. |
| Ligand Exchange | Chiral ligands (e.g., proline) complexed with metal ions (e.g., Cu²⁺) [74] | Formation of diastereomeric complexes between the metal ion, the chiral selector, and the enantiomers, primarily for separations of amino acids and hydroxych acids. |
Method Development and Screening Strategies
Developing a robust chiral HPLC method requires a systematic screening approach due to the difficulty in predicting which CSP and mobile phase will be successful for a given molecule. A common strategy involves primary screening with a limited set of complementary CSPs and mobile phases to maximize the success rate while minimizing time and resource expenditure [71]. A standard screening protocol might use three to four polysaccharide-based columns (e.g., the immobilized set of Chiralpak IA, IB, IC or the coated set of Chiralpak AD-H, AS-H, Chiralcel OD-H, OJ-H) with a set of four solvent mixtures in normal-phase mode (e.g., heptane/ethanol, heptane/isopropanol, heptane/ethyl acetate, and heptane/dichloromethane with additives like diethylamine or trifluoroacetic acid) [71]. This approach can achieve enantiorecognition for over 90% of racemates.
For molecules that are polar, ionic, or destined for LC-MS analysis, reversed-phase (RP) conditions are preferred. In RP, the mobile phase is typically a water-miscible organic solvent (e.g., methanol or acetonitrile) combined with an aqueous buffer. The pH of the buffer is critical and is chosen to suppress the ionization of acidic or basic analytes, often using formic acid for acids (pH ~2-2.5) or ammonium bicarbonate for bases (pH ~9) to ensure MS compatibility [71].
Supercritical Fluid Chromatography (SFC) for Chiral Separation
Supercritical Fluid Chromatography (SFC) is a chromatographic technique that uses supercritical carbon dioxide (sCO₂) as the primary component of the mobile phase. sCO₂, which exists above its critical temperature (31.1°C) and pressure (73.8 bar), exhibits properties of both a gas and a liquid: low viscosity and high diffusivity like a gas, and solvating power like a liquid [75]. These properties make SFC a powerful technique for chiral separations, offering several advantages over traditional HPLC.
Advantages of SFC
- Enhanced Efficiency and Speed: The low viscosity of sCO₂ allows for the use of higher flow rates without generating excessive backpressure, leading to faster separations. The high diffusivity of analytes in sCO₂ improves mass transfer, resulting in higher column efficiency [75] [73].
- Environmental and Economic Benefits: sCO₂ is non-toxic, non-flammable, and readily available, making SFC a "greener" alternative to normal-phase HPLC, which often employs large quantities of hexane and other hazardous solvents. The cost of sCO₂ is also lower than that of high-purity organic solvents [75].
- Ease of Fraction Recovery: In preparative applications, the recovery of purified compounds is simplified because CO₂ evaporates spontaneously upon depressurization, leaving behind a highly concentrated product [75].
- Versatility in Modifiers: While alcohols like methanol and ethanol are the most common co-solvents ("modifiers") in SFC, the technique—especially when used with immobilized CSPs—is compatible with a wider range of solvents, including tetrahydrofuran (THF) and methyl tert-butyl ether (MtBE), which can unlock unique selectivities [71].
SFC Method Development
Method development in SFC typically involves screening CSPs similar to those used in HPLC, with the mobile phase consisting primarily of sCO₂ and a polar organic modifier. Ethanol and methanol are the preferred initial co-solvents due to their strong eluting power and environmental profile [71] [75]. The modifier percentage is a key parameter for controlling retention and selectivity. As in HPLC, additives like diethylamine or trifluoroacetic acid are used to improve peak shape for ionizable compounds. The backpressure and temperature are additional parameters that can be optimized to fine-tune the separation.
Advanced Materials and Cutting-Edge Innovations
High-Efficiency Chiral Stationary Phases
Recent advancements in particle technology have significantly improved the efficiency of CSPs, which has historically lagged behind that of achiral columns.
- Sub-2-Micron Fully Porous Particles (FPPs): The use of small, narrow particle size distribution (NPSD) FPPs (e.g., 1.9 µm) follows the principles of UHPLC, leading to a dramatic increase in efficiency. For example, a separation of propranolol on a teicoplanin-based 1.9-µm NPSD CSP showed a more than four-fold increase in plate count compared to a standard 5-µm column, effectively doubling the resolution [73].
- Superficially Porous Particles (SPPs): SPPs (also known as core-shell particles), typically around 2.7 µm in diameter, offer high efficiency with lower backpressure than sub-2-µm FPPs. SPP-based CSPs for cyclodextrins and cyclofructans have demonstrated about three times the efficiency of commercial 5-µm FPP columns [73]. Their high permeability makes them ideal for ultrafast chiral separations, enabling resolutions in the sub-second to 40-second range [73].
The Rise of AI and Predictive Modeling in Method Development
The complexity of chiral method development, with its interdependent parameters, is being addressed by artificial intelligence (AI) and machine learning (ML). Emerging tools use digital twins and mechanistic modeling to autonomously optimize HPLC methods with minimal experimentation [76]. Furthermore, Quantitative Structure-Enantioselective Retention Relationship (QSERR) models are being developed. These models use both achiral and chiral molecular descriptors derived from molecular dynamics simulations to predict enantioselective behavior, retention, and even elution order on polysaccharide-based CSPs, paving the way for rational, computer-assisted method design [76].
Preparative-Scale Applications
The ultimate goal of analytical screening is often to develop a method for preparative-scale purification, which is crucial for providing pure enantiomers for toxicological studies, clinical trials, and as reference standards. Both HPLC and SFC are used for this purpose. Preparative SFC, in particular, has gained prominence due to its higher productivity (faster cycle times and higher throughput) and significantly reduced solvent consumption, which lowers operational costs and environmental impact [75]. The easy removal of CO₂ after collection also results in a more concentrated and easily isolated product.
Experimental Protocols and Workflows
A Standard Workflow for Analytical Chiral Screening
Diagram Title: Chiral Method Development Workflow
Detailed Protocol: Enantiomer-Specific Analysis of Underivatized Amino Acids
This protocol, adapted from recent research, details the isolation of individual L- and D-amino acids for advanced isotopic analysis, demonstrating a highly specialized application of chiral HPLC [72].
- Objective: To chromatographically isolate underivatized L- and D-enantiomers of alanine and valine for subsequent radiocarbon analysis.
- Materials and Equipment:
- HPLC System: Agilent 1290 HPLC with Diode Array Detector (DAD) and Charged Aerosol Detector (CAD).
- Chiral Column: Dr. Maisch GmbH Reprosil CHIRAL AA (250 x 4.6 mm, 8 µm) with a teicoplanin aglycone selector.
- Cleanup Column: SIELC Primesep A mixed-mode reversed-phase cation exchange column (50 x 10 mm, 5 µm).
- Mobile Phases: For chiral separation: 85% Methanol, 15% Water (isocratic). For cleanup: (A) Water with 0.1% Trifluoroacetic acid (TFA), (B) Acetonitrile with 0.1% TFA.
- Standards: L-, D-, and racemic mixtures of alanine and valine.
- Procedure:
- Chiral Separation:
- Dissolve the amino acid sample in pure water.
- Inject onto the Reprosil CHIRAL AA column.
- Run isocratically with 85% MeOH / 15% H₂O at 1.5 mL/min and 20°C.
- Monitor at 214 nm using DAD.
- Collect fractions based on retention windows: L-alanine (5-14 min), D-alanine (15-32 min), L-valine (4-8 min), D-valine (10-20 min).
- Dry collected fractions under a gentle stream of N₂ or Ar.
- Cleanup Step:
- Redissolve the dried enantiomer in 0.1 M HCl.
- Inject onto the Primesep A column.
- Run isocratically at 95% A / 5% B at 1.5 mL/min.
- Collect the enantiomer again (Alanine: 6-11 min; Valine: 12-19 min).
- Dry down the purified fraction.
- Purity and Composition Check:
- Verify chromatographic purity using an orthogonal method (e.g., HPLC-CAD with a graphite carbon column).
- Measure the Carbon-to-Nitrogen (C/N) ratio using a nano-Elemental Analyzer/Isotope Ratio Mass Spectrometer (EA/IRMS) to confirm the absence of carbon contamination from the process.
- Chiral Separation:
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Chiral Chromatography
| Item | Function and Importance |
|---|---|
| Polysaccharide-Based CSPs (Chiralpak IA/IB/IC, AD/AS, Chiralcel OD/OJ) | Workhorse columns for chiral screening; coated and immobilized versions offer broad applicability across NP, RP, and SFC modes [71]. |
| Macrocyclic Glycopeptide CSPs (Vancomycin, Teicoplanin) | Versatile for RP and HILIC separations, especially effective for amino acids, peptides, and other polar compounds [73] [72]. |
| Carbon Dioxide (SFC Grade) | The primary mobile phase component in SFC; its supercritical state provides the unique solvating power and kinetic properties that define the technique [75]. |
| HPLC/SFC Grade Modifiers & Additives (Ethanol, Methanol, Isopropanol, Diethylamine, Trifluoroacetic Acid) | Co-solvents and additives are critical for fine-tuning retention, selectivity, and peak shape. High purity is essential for reproducibility and low background noise [71]. |
| Buffers for Reversed-Phase (Ammonium Bicarbonate, Formic Acid) | Enable control of mobile phase pH to suppress analyte ionization, ensuring reproducible retention and MS compatibility [71]. |
The chromatographic separation of enantiomers via HPLC and SFC has matured into a powerful and indispensable suite of technologies in modern organic and pharmaceutical research. Driven by the critical importance of stereochemical purity in biological systems, the field has evolved from relying on a handful of columns to possessing a sophisticated toolkit of high-efficiency CSPs, green and rapid SFC methodologies, and predictive computational models. The choice between HPLC and SFC, and within them the selection of mode and stationary phase, is guided by the physicochemical properties of the analyte and the ultimate goal of the analysis, whether it is analytical quantification or preparative-scale isolation. As therapeutic agents become more complex and the demand for efficiency and sustainability grows, the continued innovation in chiral separation technologies—spearheaded by new materials, AI-driven optimization, and greener processes—will remain a cornerstone of drug development and molecular discovery.
Asymmetric Synthesis Strategies for Enantioselective Drug Production
The three-dimensional structure of a drug molecule is a critical determinant of its biological activity. Stereochemistry, particularly chirality, underpins the interactions between a pharmaceutical compound and its biological target, which is itself a chiral environment [77]. Asymmetric synthesis, also known as enantioselective synthesis, comprises the set of methodologies used to preferentially produce one enantiomer of a chiral molecule over the other [78]. This technical guide details the core strategies, methodologies, and applications of asymmetric synthesis within modern drug production, framed within the essential context of stereochemistry and isomerism.
The significance of enantioselective synthesis in pharmaceuticals was starkly highlighted by the thalidomide tragedy, where one enantiomer provided therapeutic effect while the other caused severe birth defects [77]. This understanding, combined with evolving regulatory landscapes, has firmly established asymmetric synthesis as a cornerstone of modern drug development. The global market for chiral technology, valued at US$8.6 billion in 2024 and projected to reach US$10.7 billion by 2030, reflects the critical importance of these methods in producing enantiomerically pure pharmaceuticals [79].
Core Concepts of Stereochemistry in Drug Action
Isomerism and Chirality
Isomers are molecules with the same molecular formula but different structural arrangements, broadly categorized as either constitutional isomers or stereoisomers [54].
- Constitutional Isomers: Differ in the connectivity of their atoms (e.g., 1-propanol vs. 2-propanol) [80].
- Stereoisomers: Share the same atomic connectivity but differ in the spatial orientation of their atoms. This category includes enantiomers and diastereomers [54].
Enantiomers are a pair of stereoisomers that are non-superimposable mirror images of each other [80]. A molecule is chiral if it lacks an internal plane of symmetry and thus can exist as a pair of enantiomers. The central role of chirality in drug action arises because biological systems—composed of chiral building blocks like L-amino acids and D-sugars—interact with each enantiomer of a drug differently. Often, one enantiomer (the eutomer) possesses the desired therapeutic activity, while its mirror image (the distomer) may be inactive, exhibit a different pharmacological profile, or be toxic [77] [81].
Regulatory and Commercial Drivers
Regulatory agencies, including the FDA and European Medicines Agency (EMA), strongly favor the development of single-enantiomer drugs over racemic mixtures [81]. This preference is driven by the need for improved drug safety, efficacy, and predictability. Recent analysis shows that from 2013 to 2022, the EMA has not approved a single racemate since 2016, while the FDA has averaged only one racemic approval per year [81]. This regulatory environment, coupled with the potential for extended patent protection via "chiral switches" (e.g., the development of escitalopram from citalopram), creates a powerful incentive for the pharmaceutical industry to master enantioselective synthesis [7].
Foundational Strategies for Asymmetric Synthesis
The primary goal of asymmetric synthesis is to convert an achiral starting material into a chiral product with a high preference for one enantiomer, quantified by the enantiomeric excess (e.e.). The three main strategic approaches are outlined below.
The Chiral Pool Approach
This strategy utilizes readily available, enantiopure natural products—such as amino acids, sugars, or terpenes—as building blocks to introduce chirality into a target molecule [77]. The stereochemical information inherent in these "pool" molecules is transferred to the synthetic intermediate or final drug substance.
- Example - Lifitegrast (2016): The synthesis of Lifitegrast, a treatment for dry eye disease, begins with the commercially available chiral substrate 3-bromo-l-phenylalanine (A1). The existing stereocenter in this amino acid is used to construct the final active pharmaceutical ingredient (API) in 10 steps with an 88% yield [77].
Chiral Resolution
Chiral resolution is a process for separating the enantiomers of a racemic mixture. This method does not create new chirality but is a practical way to obtain an enantiomerically pure compound from a racemate.
- Techniques: The most common technique is diastereomeric salt formation, where the racemic mixture is reacted with an enantiopure chiral acid or base to form two diastereomeric salts. These salts possess different physical properties (e.g., solubility) and can be separated by crystallization. Chiral Chromatography using High-Performance Liquid Chromatography (HPLC) with Chiral Stationary Phases (CSPs) is another highly effective analytical and preparative method [81].
Asymmetric Synthesis
This is the most direct and economically favorable approach, where chirality is introduced during a synthetic step using a chiral auxiliary, reagent, or catalyst [77]. The following table summarizes key methodologies.
Table 1: Core Methodologies in Asymmetric Synthesis
| Methodology | Key Feature | Example Application in Drug Synthesis |
|---|---|---|
| Chiral Auxiliary | A temporary chiral unit controls stereochemistry in one or more steps and is later removed. | Netarsudil (2017): An Evans oxazolidinone (A15) was used as a chiral auxiliary to install the essential stereochemistry, which was later removed with hydrogen peroxide [77]. |
| Chiral Reagent | A stoichiometric chiral reactant induces asymmetry. | Pemafibrate (2017): The chiral fragment was synthesized from enantiopure (S)-2-hydroxybutyrolactone (A7) [77]. |
| Asymmetric Catalysis | A sub-stoichiometric chiral catalyst promotes the formation of one enantiomer. | Lorlatinib (2018): A key chiral alcohol intermediate (A25) was constructed via enantioselective reduction using a biocatalyst [77]. |
The Three Pillars of Catalytic Asymmetric Synthesis
Catalytic asymmetric synthesis is the most efficient and widely used method for enantioselective production on an industrial scale. It relies on three primary classes of catalysts, often termed the "three pillars" of asymmetric catalysis [78].
Transition-Metal Catalysis
This method employs transition metals (e.g., Rh, Pd, Ni, Cu) complexed with chiral ligands to create a chiral environment around the metal center. This environment directs the approach of reactants, leading to enantioselective bond formation [82] [78].
- Recent Advancements: A 2025 review highlights several breakthroughs, including Ni-catalyzed hydroalkylation of fluoroalkenes for synthesizing fluorinated compounds with adjacent chiral centers, and Cu-catalyzed dearomative cyclopropanation of indole-diynes to access complex indoline structures [82].
- Industrial Impact: The work of Knowles, Noyori, and Sharpless (Nobel Prize, 2001) on asymmetric hydrogenation and oxidation using metal catalysts revolutionized the industrial production of L-DOPA and other chiral pharmaceuticals [78].
Biocatalysis
Biocatalysis harnesses the power of enzymes, nature's own chiral catalysts, to perform highly enantioselective transformations under mild conditions [78].
- Mechanism: Enzymes provide a highly tailored, three-dimensional active site that perfectly accommodates only one enantiomer of a substrate or transition state.
- Engineering & Applications: Modern protein engineering and directed evolution allow scientists to optimize the enantioselectivity and substrate scope of enzymes for non-natural reactions. An exemplary application is the synthesis of a key piperidine intermediate (A22) for the cancer drug Niraparib (2017), which uses a transaminase enzyme (ATA-302) co-catalyzed by pyridoxal-5-phosphate (PLP) [77].
Organocatalysis
Organocatalysis employs small organic molecules, devoid of metals, to catalyze enantioselective reactions. These catalysts often function by forming transient covalent bonds or through hydrogen-bonding interactions with substrates [78].
- Mechanism: A prominent mode of action involves iminium ion catalysis, where a chiral secondary amine (e.g., proline or a MacMillan catalyst) reacts with an α,β-unsaturated aldehyde to form a chiral iminium ion intermediate. This activation lowers the LUMO energy and shields one face of the molecule, leading to enantioselective attack by a nucleophile.
- Significance: The development of organocatalysis by Benjamin List and David MacMillan (Nobel Prize, 2021) provided a powerful, stable, and low-toxicity catalytic platform that complements metal and enzyme catalysis [78].
Table 2: Comparison of the Three Pillars of Asymmetric Catalysis
| Feature | Transition-Metal Catalysis | Biocatalysis | Organocatalysis |
|---|---|---|---|
| Typical Catalysts | Rh, Pd, Ni, Ru complexes with chiral ligands (e.g., BINAP, Salox) | Enzymes (e.g., ketoreductases, transaminases, P450s) | Small organic molecules (e.g., proline, cinchona alkaloids) |
| Key Strength | Broad substrate scope, high functional group tolerance | Unmatched selectivity and green credentials | Low toxicity, air/water stability, ease of handling |
| Reaction Types | Hydrogenation, C-C cross-coupling, C-H functionalization | Reductions, oxidations, C-N bond formation | Aldol, Michael addition, cyclopropanation |
| Scalability | Excellent, well-established in industry | Excellent for specific, optimized processes | Very good, often simple reaction setups |
Experimental Protocols and Workflows
Protocol: Biocatalytic Reduction for Chiral Alcohol Synthesis
This protocol describes a generalized procedure for the enantioselective reduction of a prochiral ketone to a chiral alcohol using a ketoreductase enzyme, analogous to the synthesis of the Lorlatinib intermediate [77].
- Step 1: Reaction Setup: In a suitable buffer (e.g., phosphate buffer, pH 7.0), combine the prochiral ketone substrate (1.0 equiv), a nicotinamide cofactor (NADP⁺, catalytic amount), and the ketoreductase enzyme (1-5 mg/mL). Include a cofactor recycling system, such as isopropanol (2.0 equiv) or a glucose/glucose dehydrogenase pair, to regenerate the active NADPH form.
- Step 2: Process Execution: Stir the reaction mixture at a controlled temperature (25-35°C) and monitor the reaction progress by chiral HPLC or GC until >95% conversion is achieved.
- Step 3: Work-up and Isolation: Extract the product with an organic solvent (e.g., ethyl acetate). Purify the chiral alcohol by flash chromatography or distillation. Determine the enantiomeric excess (e.e.) by chiral HPLC analysis.
Protocol: Chiral HPLC for Enantiomeric Excess Determination
The determination of e.e. is a critical analytical step in asymmetric synthesis. Chiral HPLC is the gold standard for this purpose [81].
- Step 1: Method Development: Select an appropriate Chiral Stationary Phase (CSP). Common CSPs include Chiralpak IA/IB/IC (amylose-based) or Chiralcel OD/OJ (cellulose-based) columns. Prepare a dilute solution of the racemic mixture (if available) and the enantiomerically enriched sample.
- Step 2: Mobile Phase Optimization: Use a normal-phase solvent system (e.g., hexane/isopropanol) or a reverse-phase system (acetonitrile/water with 0.1% acid) depending on the analyte. Systematically vary the ratio of organic modifiers (e.g., 5-20% isopropanol in hexane) and the column temperature to achieve baseline resolution (Rₛ > 1.5) of the enantiomer peaks.
- Step 3: Quantification: Inject the enantiomerically enriched sample. The e.e. is calculated using the formula: e.e. (%) = ([A] - [B]) / ([A] + [B]) × 100%, where [A] and [B] are the peak areas of the major and minor enantiomers, respectively.
Workflow Visualization
The following diagram illustrates a generalized decision-making workflow for selecting an asymmetric synthesis strategy in drug development.
Diagram 1: Strategy Selection Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of asymmetric synthesis requires a suite of specialized reagents, catalysts, and analytical tools.
Table 3: Key Research Reagent Solutions for Asymmetric Synthesis
| Reagent/Material | Function | Example/Category |
|---|---|---|
| Chiral Ligands | Bind to transition metals to form chiral catalysts, controlling the enantioselectivity of the reaction. | BINAP, Salox Ligands, Josiphos, Pyridinebisoxazoline [82]. |
| Chiral Organocatalysts | Small organic molecules that catalyze reactions through covalent or non-covalent interactions. | L-Proline, Cinchona Alkaloids, MacMillan Imidazolidinone Catalysts [78]. |
| Enzymes (Biocatalysts) | Provide high enantio- and regioselectivity for specific transformations under mild conditions. | Ketoreductases (KREDs), Transaminases (ATAs), P450 Monooxygenases [77] [78]. |
| Chiral Stationary Phases (CSPs) | Used in HPLC for analytical and preparative separation of enantiomers to determine e.e. and isolate pure isomers. | Cellulose-based (Chiralcel OD/OJ), Amylose-based (Chiralpak IA/AD) [81]. |
| Chiral Auxiliaries | Temporary chiral units covalently attached to control stereochemistry in one or more steps, then removed. | Evans Oxazolidinones, Oppolzer's Sultams [77]. |
| Chiral Pool Building Blocks | Commercially available, enantiopure natural products used as starting materials to transfer chirality. | L-amino acids, D-sugars, Lactic acid, (S)-2-hydroxybutyrolactone [77]. |
Asymmetric synthesis has evolved from a academic challenge to an indispensable component of modern pharmaceutical production. The drive for enantiomerically pure drugs, fueled by regulatory requirements and the pursuit of safer, more efficacious therapies, has cemented its role in the drug development pipeline. The continued innovation in transition-metal catalysis, biocatalysis, and organocatalysis provides medicinal chemists with a powerful and versatile toolkit. Future progress will likely be driven by the integration of these methods, the application of machine learning for catalyst design and reaction optimization, and a growing emphasis on green chemistry principles to make enantioselective processes more sustainable. A deep understanding of stereochemistry and the strategic application of asymmetric synthesis are, and will remain, fundamental to successful drug discovery and development.
Stereoselectivity, the ability of a chemical reaction to preferentially produce one stereoisomer over another, is a cornerstone of modern organic synthesis. Its control is critical across scientific disciplines, from the development of chiral pharmaceuticals to the design of advanced energetic materials. This phenomenon arises from the differential interaction of stereoisomers with chiral environments, whether in the active site of an enzyme, the pocket of a synthetic catalyst, or a crystalline lattice. The spatial arrangement of atoms directly dictates a molecule's biological activity, physical properties, and chemical behavior [83]. Within the broader context of stereochemistry and isomerism research, understanding and predicting stereoselectivity represents a fundamental challenge, driving the development of increasingly sophisticated theoretical models and experimental techniques. This guide provides an in-depth examination of the mechanisms, controlling factors, and state-of-the-art tools for analyzing and predicting stereoselective outcomes, framed for researchers and drug development professionals.
Fundamental Concepts and Historical Models
Stereoselectivity is fundamentally governed by the energy difference between diastereomeric transition states leading to stereoisomeric products. The degree of this selectivity is quantified as the enantiomeric ratio (e.r.) or the enantioselectivity, expressed as ΔΔG‡ = -RT ln(e.r.), where ΔΔG‡ represents the difference in activation energies for the pathways leading to each enantiomer [4]. The evolution of strategies for controlling stereogenic centers can be divided into three major approaches: substrate control, which utilizes inherent chirality in the starting material; chiral stoichiometric reagent control; and catalyst control (asymmetric catalysis), which uses a chiral catalyst in substoichiometric amounts to provide kinetic discrimination between enantiomers through diastereomeric transition state structures [84].
Early understanding was built upon qualitative models derived from chemical intuition and meticulously designed experiments. These models, represented as simple molecular projections, provided a practical framework based on steric and electronic effects.
Table 1: Classical Stereochemical Models for Carbonyl Compounds
| Name of Model/Reaction | Substrate Type | Use Case & Key Principle |
|---|---|---|
| Cram's Rule [84] | α-Chiral carbonyls | Empirical model for nucleophilic additions; minimizes steric contacts by placing the bulkiest substituent (RL) anti to the carbonyl. |
| Felkin-Anh Model [84] | α-Chiral carbonyls | Improved model using a staggered conformation with RL perpendicular to the carbonyl; nucleophile approaches from the less hindered side. |
| Cram's Chelation Rule [84] | α-Chiral carbonyls | Applies when a chelating metal ion coordinates to the carbonyl oxygen and an α-substituent, altering the preferred conformation. |
| Zimmerman-Traxler Model [84] | Enolates and carbonyls | Proposes a pseudo-six-membered ring transition state for aldol reactions, favoring the chair-like geometry with minimal 1,3-diaxial interactions. |
The Felkin-Anh and Zimmerman-Traxler models remain robust frameworks for understanding diastereoselectivity in fundamental reactions. However, their limitations prompted the development of more advanced, quantitative tools capable of handling complex molecular interactions [84].
Figure 1: The evolution of predictive tools for stereoselectivity, from simple qualitative pictures to complex machine learning models [84].
Modern Predictive Tools and Computational Methods
The challenge of quantitatively predicting stereoselectivity, particularly for complex catalytic systems, has been addressed by significant advances in computational chemistry and data science.
Quantum Mechanical Calculations
Density Functional Theory (DFT) calculations have become indispensable for elucidating the mechanism and origin of selectivities. For instance, a recent DFT study on cobalt-catalyzed C–H functionalization of arylphosphinamide revealed a stepwise mechanism where C–H cleavage and alkyne insertion were stereoselectivity-determining. The study found that pronounced S-selectivity originated from a larger number of stabilizing non-covalent interactions in the lower-energy transition state [85]. Such computational insights provide an atomistic understanding that guides the rational design of more selective catalysts.
Machine Learning and Data Science
Machine learning (ML) has emerged as a powerful complement to traditional computational methods, especially for navigating vast chemical spaces and predicting reaction outcomes like enantioselectivity (ΔΔG‡) [84] [4]. ML models can capture complex, non-linear relationships between molecular features and selectivity that are difficult to model with linear regression or intuition alone.
Table 2: Machine Learning Approaches for Predicting Stereoselectivity
| Method | Key Application & Advantage | Representative Algorithm(s) |
|---|---|---|
| Composite ML Methods [4] | Uses Bayesian optimization and permutation importance tests to select the best model (e.g., RF, SVR, LASSO) for a new reaction based on its features via Gaussian Mixture Models. | Random Forest (RF), Support Vector Regression (SVR), LASSO |
| Stereochemistry-Aware Generative Models [23] | Molecular generation explicitly including stereochemical information (R/S, E/Z), optimizing stereochemistry-sensitive properties in drug design. | Reinforcement Learning (e.g., REINVENT), Genetic Algorithms (e.g., JANUS) |
| Deep Learning & Graph Neural Networks [4] | Predicting solvation Gibbs free energy and other quantum mechanical properties related to transition state energies from molecular structure. | Deep Neural Networks (DNN), Graph Neural Networks (GNN) |
A key development is the creation of stereochemistry-aware generative models for molecular design. These models explicitly incorporate stereochemical information (e.g., R/S and E/Z configuration) into string-based representations like SMILES and SELFIES. Benchmarking shows that these models perform as well as or better than stereochemistry-unaware models on tasks sensitive to 3D structure, such as optimizing binding affinity or optical activity, despite the increased complexity of the chemical space they must explore [23].
Experimental Protocols and Analytical Techniques
Verifying stereoselective outcomes and analyzing stereoisomers requires precise and sensitive chiral analytical techniques.
Protocol for Chromatographic Enantioseparation
Principle: High-performance liquid chromatography (HPLC) using a Chiral Stationary Phase (CSP) is a direct and widely used method for separating and quantifying enantiomers [83]. The CSP provides a chiral environment that differentially interacts with each enantiomer, leading to separation.
Detailed Methodology:
- Column Selection: Select an appropriate CSP (e.g., polysaccharide-, cyclodextrin-, or Pirkle-type) based on the analyte's structure and literature precedent.
- Sample Preparation: Dissolve the racemic mixture or reaction product in a suitable solvent (e.g., methanol, acetonitrile, or the mobile phase) at a known concentration. Filter through a 0.45 μm or 0.22 μm syringe filter to remove particulates.
- Mobile Phase Optimization: Begin with a standard mobile phase (e.g., hexane/isopropanol). Systematically vary the composition (e.g., 90:10 to 70:30 hexane/IPA) to achieve baseline separation of enantiomer peaks. For more polar compounds, reverse-phase conditions (water/acetonitrile with modifiers) may be necessary.
- Instrumental Parameters:
- Flow Rate: Typically 0.5 - 1.0 mL/min for analytical columns (4.6 mm diameter).
- Detection: Use a UV-Vis detector at the analyte's λmax or a generic wavelength (e.g., 214 nm, 254 nm).
- Injection Volume: 1 - 20 μL, depending on concentration and detector sensitivity.
- Column Temperature: Hold constant (e.g., 25 °C) or optimize for better separation.
- Data Analysis: Identify enantiomer peaks by comparing retention times with pure standards if available. Calculate the enantiomeric ratio (e.r.) or enantiomeric excess (e.e.) from the peak areas: e.e. (%) = [|Area₁ - Area₂| / (Area₁ + Area₂)] * 100.
Advanced and Complementary Techniques
- Capillary Electrophoresis (CE): Uses chiral selectors added to the background electrolyte. It offers high efficiency and different selectivity compared to HPLC, making it a valuable orthogonal method [83].
- Mass Spectrometry (MS) as Detector: Coupling HPLC or CE with MS provides high sensitivity and specificity for identifying enantiomers, especially in complex matrices like biological fluids. However, matrix effects can remain a challenge [83].
- Nuclear Magnetic Resonance (NMR): NMR can be used for chiral analysis by employing chiral solvating agents or derivatizing agents, which cause diastereomeric interactions detectable in the NMR spectrum [83].
Table 3: Key Research Reagent Solutions for Stereoselective Analysis
| Reagent / Material | Function in Stereoselectivity Research |
|---|---|
| Chiral Stationary Phases (CSPs) [83] | The core of chiral HPLC and SFC; provides a chiral environment for the direct separation and analysis of enantiomers. |
| Chiral Solvating Agents (CSAs) [83] | Used in NMR spectroscopy to differentiate enantiomers by forming transient diastereomeric complexes that give distinct chemical shifts. |
| Chiral Catalysts (e.g., CPAs) [4] | Small molecules (e.g., BINOL-derived phosphoric acids) used in asymmetric synthesis to induce enantioselectivity in the reaction product. |
| Deuterated Solvents | Essential for NMR-based chiral analysis, allowing for the study of molecular structure and configuration in solution. |
| Chiral Derivatization Agents [83] | Convert enantiomers into diastereomers via chemical reaction, enabling separation and analysis on standard (achiral) chromatographic systems. |
Figure 2: Standard workflow for analyzing stereoselective reaction outcomes using chiral chromatography.
Impact and Applications in Research and Industry
The implications of stereoselectivity are profound and far-reaching, directly impacting research and development outcomes.
Pharmaceutical Sciences and Drug Metabolism
In drug metabolism, stereoselectivity is a dominant factor due to the chiral nature of enzymes. This leads to substrate stereoselectivity, where enantiomers are metabolized at different rates, and product stereoselectivity, where a single substrate forms stereoisomeric metabolites preferentially [83]. A classic example is the proton pump inhibitor omeprazole. Its (R)- and (S)-enantiomers are metabolized by different cytochrome P450 enzymes (CYP2C19 and CYP3A4, respectively), leading to different clearance rates and oral bioavailability. This understanding led to the development of esomeprazole, the (S)-enantiomer, as a more efficacious single-enantiomer drug [83]. The tragic case of thalidomide and the differential cardiac effects of (R)- and (S)-methadone further underscore the critical importance of stereochemical assessment in drug development and therapeutic drug monitoring [23] [83].
Materials Science
Stereochemistry is also a critical design parameter in materials science. Recent research on cage-like 3D energetic materials based on a 2,4,10-trioxaadamantane backbone demonstrated that diastereomers, despite having identical molecular formulas and functional groups, exhibit measurable differences in density, stability, and detonation performance. This "stereochemical editing" presents a novel and efficient strategy for fine-tuning the properties of high-energy density materials beyond the traditional approach of adding more explosive functional groups [86].
The pursuit of controlling and predicting stereoselectivity has driven the evolution of tools from simple hand-drawn models to sophisticated integrations of quantum chemistry and machine learning. This progression reflects a broader paradigm shift in organic chemistry towards data-driven, predictive science. For researchers and drug development professionals, a multidisciplinary toolkit is now essential. This toolkit combines foundational stereochemical principles, modern computational simulations to unravel mechanistic details, ML models to navigate complex reaction spaces, and robust analytical protocols for unambiguous verification. As stereochemistry-aware molecular generation and predictive models continue to mature, they promise to accelerate the rational design of chiral molecules with optimal properties, solidifying the central role of stereoselectivity in the future of chemical synthesis, pharmaceutical research, and advanced materials design.
Bioanalytical Method Development for Isomer Quantification
In the realms of pharmaceutical development and clinical science, the precise quantification of molecular isomers is not merely an analytical exercise but a fundamental prerequisite for ensuring drug safety and efficacy. Isomers, molecules with identical atomic formulas but differing spatial arrangements, are ubiquitous; they are present in naturally occurring compounds such as steroids and sugars, as well as in synthetically derived pharmaceuticals [87]. The physiological functions of different stereoisomers can diverge dramatically. A paramount example is the drug thalidomide, where one enantiomer functioned as a sedative while its mirror image acted as a potent teratogen, leading to severe birth defects [88]. This historical lesson underscores a critical reality: in drug development, chiral molecules represent distinct chemical entities [32].
For researchers and drug development professionals, this imposes a significant analytical challenge. The biological properties of chiral molecules are inextricably linked to their three-dimensional structure, mandating the use of robust bioanalytical methods that can reliably differentiate and quantify these stereoisomers in complex biological matrices [32]. This guide provides an in-depth technical framework for developing and validating such methods, contextualized within the broader principles of stereochemistry and isomerism in organic molecules research.
Stereochemical Foundations and Bioanalytical Implications
Fundamental Types of Isomers
Isomers are broadly categorized based on their structural and spatial characteristics, each with profound implications for bioanalysis:
- Constitutional Isomers: These possess the same molecular formula but differ in the connectivity of their atoms (i.e., different bond connectivities) [88].
- Stereoisomers: These share the same atomic connectivity but differ in the three-dimensional orientation of their atoms in space [88]. Stereoisomers are further subdivided into:
A molecule is deemed chiral if it is not identical to its mirror image; the most common source of chirality is the presence of one or more chiral centers—typically a tetrahedral carbon atom bonded to four different substituents [88]. The absolute configuration of a chiral center, denoted as R or S, defines its precise spatial arrangement [32].
The Pharmacological Significance of Stereoisomers
The necessity for isomer-specific bioanalytical methods stems from the distinct biological behaviors of stereoisomers. In a chiral environment, such as the human body where proteins, enzymes, and receptors are themselves chiral, enantiomers are perceived as different molecules [88]. They can exhibit null, similar, different, or even opposite therapeutic activities [32]. Consequently, the incorrect stereochemical assignment or the use of a racemic mixture (a 50/50 mixture of enantiomers) can lead to unpredictable and potentially severe clinical consequences [32]. The US Food and Drug Administration (FDA) and other regulatory bodies therefore require rigorous stereochemical characterization of chiral active pharmaceutical ingredients (APIs) [89].
Analytical Techniques for Isomer Separation and Detection
Overcoming the challenge of isomer quantification requires a synergistic combination of high-resolution separation techniques and selective detection methods.
Chromatographic Separation Strategies
Chromatography is the cornerstone of isomer separation, with the choice of stationary phase being paramount:
- Chiral Stationary Phases (CSPs): These columns contain a chiral selector that interacts differentially with enantiomers, enabling their direct separation. For instance, a Lux-i-Amylose-3 chiral column was successfully used to achieve baseline separation of Paroxetine enantiomers using a mobile phase of acetonitrile and acetate buffer [89].
- Specialized Reversed-Phase Columns: For diastereomers or geometric isomers, specialized columns can provide enhanced resolution. A C30 carotenoid column, for example, has been employed to separate all-trans-lycopene from its various cis isomers using a gradient solvent system [90].
Detection and Hyphenated Techniques
Following separation, detection must provide the specificity and sensitivity needed for bioanalysis.
- Mass Spectrometry (MS): While isomers have identical mass-to-charge ratios, tandem mass spectrometry (MS/MS) can generate distinct fragmentation patterns. For example, lycopene isomers produced a unique fragment of m/z 467 from a molecular ion of m/z 536, allowing selective detection via multiple reaction monitoring (MRM) [90]. Advanced techniques like ion mobility spectrometry (IMS) can further separate ions in the gas phase based on their size and shape, providing an additional dimension of separation [87].
- Chiroptical Methods: Techniques such as Electronic Circular Dichroism (ECD) and Vibrational Circular Dichroism (VCD) are crucial for determining absolute configuration (AC) and conformation, especially when X-ray crystallography is not feasible [32]. These methods measure the differential interaction of a chiral molecule with left- and right-circularly polarized light.
- Nuclear Magnetic Resonance (NMR) Spectroscopy: NMR provides definitive structural information, including the ability to distinguish between isobaric compounds and positional isomers, which MS often cannot [91]. Its primary limitation in bioanalysis is relatively low sensitivity compared to MS [91].
Integrated LC-MS-NMR platforms represent the pinnacle of comprehensive characterization, combining the separation power of LC with the structural elucidation capabilities of both MS and NMR [91]. Practical implementations often involve stop-flow analysis or loop collection to accommodate the longer acquisition times required for NMR [91].
Table 1: Comparison of Key Techniques for Isomer Analysis in Bioanalysis
| Technique | Principle | Key Advantages | Key Limitations | Typical LOD/LOQ |
|---|---|---|---|---|
| LC-MS/MS | Chromatographic separation followed by mass-based detection and fragmentation. | High sensitivity and specificity; Wide applicability. | Cannot distinguish isomers without prior separation or specific fragmentation. | Femtomole to picomole range [90] |
| Chiral HPLC/UFLC | Use of a chiral stationary phase for enantiomer separation. | Direct enantiomeric resolution; High preparative capacity. | Requires method optimization for each analyte; Can be expensive. | Varies with detector (e.g., MS vs. UV) |
| Circular Dichroism (CD) | Differential absorption of left- and right-circularly polarized light. | Provides AC and conformational data; Solution-state technique. | Requires quantum chemical calculations for interpretation; Signal can be complex. | Not a direct quantification technique |
| LC-NMR | Hyphenation of separation with NMR detection. | Provides definitive structural identification; Distinguishes isomers. | Inherently low sensitivity; Long acquisition times; Costly deuterated solvents often needed. | Microgram range [91] |
Method Development: A Step-by-Step Workflow
Developing a validated bioanalytical method for isomers requires a systematic and iterative approach. The following workflow outlines the critical stages.
Diagram 1: Method Development Workflow
Defining the Analytical Target and Sample Preparation
The process begins with a clear definition of the analytes (e.g., specific enantiomers, diastereomers) and the required sensitivity. Sample preparation must be optimized to ensure stability, as certain procedures like saponification can cause isomerization or degradation, as observed with lycopene [90]. Techniques such as protein precipitation, liquid-liquid extraction, or solid-phase extraction (SPE) are employed to clean up the biological matrix and pre-concentrate the analytes.
Chromatographic Optimization and Detection
The core of the method is the chiral separation. Key parameters to optimize include:
- Stationary Phase: Selection of an appropriate CSP (e.g., amylose- or cellulose-based).
- Mobile Phase: Composition (buffer pH, ionic strength, organic modifier type and percentage) and flow rate. A flow rate of 0.8 mL/min with acetonitrile and acetate buffer (70:30 v/v) was effective for Paroxetine [89].
- Temperature: Column temperature can significantly impact resolution and retention times.
Detection parameters are then tuned for the specific analytes. For MS, this involves optimizing ionization source conditions and selecting specific MRM transitions for each isomer, if available [90].
Experimental Protocol: Chiral UFLC-MS/MS Method
This protocol outlines the development of a chiral Ultra-Fast Liquid Chromatography-Mass Spectrometry (UFLC-MS/MS) method for quantifying enantiomers in plasma, based on a validated Paroxetine assay [89].
Materials and Reagents
- Analytes: Pure reference standards of individual enantiomers and the racemic mixture.
- Internal Standard (IS): A stable isotope-labeled analog of the analyte or a structurally similar compound.
- Biological Matrix: Control plasma (e.g., human, rat).
- Chemicals: HPLC-grade solvents (acetonitrile, methanol), ammonium acetate or formate, and acetic or formic acid for mobile phase preparation.
- Equipment: UFLC system, mass spectrometer with electrospray ionization (ESI), chiral column (e.g., Lux-i-Amylose-3, 250 mm x 4.6 mm, 5 µm), and a data acquisition system.
Detailed Procedure
- Standard Solution Preparation: Independently prepare separate stock solutions of each enantiomer and the IS in a suitable solvent (e.g., methanol). Dilute to working concentrations.
- Calibration Standards and QCs: Spike blank plasma with working solutions to prepare calibration standards (e.g., covering 2–10 µg/mL) and quality control (QC) samples at low, medium, and high concentrations [89].
- Sample Preparation: a. Thaw frozen plasma samples on ice and vortex. b. Aliquot a precise volume (e.g., 100 µL) into a microcentrifuge tube. c. Add a fixed volume of the IS solution. d. Precipitate proteins by adding a volume of organic solvent (e.g., 300 µL acetonitrile), vortex mix vigorously, and centrifuge at high speed (e.g., 13,000 rpm for 10 minutes). e. Transfer the clear supernatant to an autosampler vial for analysis.
- Chromatographic Conditions:
- Column: Chiral column (e.g., Lux-i-Amylose-3).
- Mobile Phase: Isocratic or gradient elution. Example: Acetonitrile and ammonium acetate buffer (10 mM, pH ~5.0) in a ratio of 70:30 (v/v) [89].
- Flow Rate: 0.8 mL/min.
- Column Temperature: Maintain constant (e.g., 25°C).
- Injection Volume: 5-10 µL.
- Mass Spectrometric Conditions:
- Ionization: ESI in positive or negative mode, optimized for the analyte.
- Data Acquisition: MRM mode. Monitor specific precursor ion → product ion transitions for each enantiomer and the IS.
- Source Parameters: Optimize desolvation temperature, gas flows, and capillary voltage.
Data Analysis
- Plot the peak area ratio (analyte/IS) against the nominal concentration of calibration standards to construct a linear regression curve.
- Use the resulting equation to calculate the concentration of enantiomers in unknown and QC samples.
Bioanalytical Method Validation
A method must be rigorously validated to demonstrate its reliability for intended use, following regulatory guidelines (e.g., US FDA) [92]. Key validation parameters are summarized below.
Table 2: Key Validation Parameters and Acceptance Criteria for Bioanalytical Methods
| Validation Parameter | Experimental Procedure | Acceptance Criteria |
|---|---|---|
| Selectivity/Specificity | Analyze blanks from at least six different sources of the biological matrix. | No significant interference at the retention times of the analyte and IS (<20% of LLOQ for analyte, <5% for IS) [92]. |
| Linearity & Range | Analyze a minimum of 6-8 non-zero calibration standards across the range. | Correlation coefficient (r) ≥ 0.99. Accuracy and precision within ±15% (±20% at LLOQ) [92] [89]. |
| Accuracy & Precision | Analyze QC samples at a minimum of three concentrations (low, medium, high) in replicates over multiple runs. | Intra- & Inter-assay Precision: %CV ≤ 15% (≤20% at LLOQ). Accuracy: %RE within ±15% (±20% at LLOQ) [92] [89]. |
| Lower Limit of Quantification (LLOQ) | Determine the lowest concentration that can be measured with acceptable accuracy and precision. | Signal-to-noise ratio ≥ 5. Accuracy and Precision within ±20% [92]. |
| Stability | Evaluate analyte stability under various conditions (bench-top, processed sample, freeze-thaw, long-term). | Deviation from nominal concentration within ±15% [92]. |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Chiral Bioanalysis
| Item | Function / Principle | Example / Note |
|---|---|---|
| Chiral HPLC Columns | Stationary phase with immobilized chiral selectors enables enantiomer separation. | Amylose- (e.g., Chiralpak AD) or cellulose-based (e.g., Chiralcel OD) columns are widely used. The Lux-i-Amylose-3 column was used for Paroxetine [89]. |
| Stable Isotope-Labeled Internal Standards (SIL-IS) | Corrects for variability in sample preparation and ionization efficiency in MS. | Deuterated (D), 13C-, or 15N-labeled analogs of the analyte. Provides best accuracy by mimicking analyte behavior. |
| High-Purity Solvents & Buffers | Mobile phase components; purity is critical for low background noise and consistent retention times. | LC-MS grade acetonitrile and methanol. Volatile buffers (ammonium acetate/formate) are preferred for LC-MS compatibility. |
| Molecularly Imprinted Polymers (MIPs) | Synthetic polymers with cavities complementary to a target molecule, used for selective sample clean-up or enrichment. | Can be designed to distinguish isomeric/isobaric species, such as sialic acid linkages [87]. |
| Immunoaffinity Reagents | Antibodies bound to a solid support used to selectively extract target analytes from complex matrices. | Used in Immunoprecipitation-MS (IP-MS) for highly specific sample preparation prior to analysis [87]. |
Application in Drug Development: A Case Study
The practical impact of chiral bioanalysis is illustrated by a pharmacokinetic study of the antidepressant Paroxetine. A validated chiral UFLC method revealed stark differences in the pharmacokinetic profiles of its enantiomers when administered as a racemate versus the individual enantiomers. The study demonstrated in vivo interconversion, with a significant amount of the R-enantiomer converting into the pharmacologically active S-enantiomer [89].
The R-enantiomer exhibited a longer half-life (4.96 hours) compared to the racemic mixture (3.41 hours), and its AUC (Area Under the Curve, a measure of systemic exposure) was higher than that of the S-enantiomer [89]. These findings are critical as they reveal that administering a racemic drug leads to a complex and changing enantiomeric ratio in the body over time, which can impact both efficacy and safety. This case powerfully advocates for the use of enantioselective analytical techniques throughout drug development to optimize formulations and dosing strategies [89].
The development of robust bioanalytical methods for the quantification of isomers is an indispensable component of modern pharmaceutical research, directly rooted in the fundamental principles of stereochemistry. As demonstrated, the distinct and often unpredictable biological activities of stereoisomers demand analytical strategies that go beyond standard quantification. Success hinges on a deep understanding of chiral separation science, coupled with the judicious application of hyphenated techniques like LC-MS and LC-NMR, and rigorous method validation. By integrating these elements, scientists can ensure the accurate characterization of chiral therapeutics, thereby paving the way for the development of safer and more effective drugs.
Stereochemistry, the study of the three-dimensional spatial arrangement of atoms in molecules, serves as a critical foundation for modern pharmaceutical science. Chirality, a geometric property where a molecule is non-superimposable on its mirror image, is a particularly essential concept in drug development [18]. Much like human hands, chiral molecules exist as two mirror-image forms called enantiomers [3]. A mixture containing equal amounts of both enantiomers is known as a racemate or racemic mixture [3]. The biological significance of chirality stems from the inherent chiral nature of living systems; proteins, enzymes, and nucleic acids are all chiral, enabling them to differentiate between enantiomers of a chiral drug [18].
This enantioselectivity means that the two mirror-image forms of a drug can exhibit dramatically different pharmacological behaviors in the body. One enantiomer (the eutomer) may possess the desired therapeutic effect, while the other (the distomer) might be inactive, contribute to side effects, or even exert an entirely different pharmacological action [93] [18]. The historical tragedy of thalidomide, where one enantiomer provided sedation and the other caused severe birth defects, starkly illustrates the profound clinical implications of stereochemistry [18]. Consequently, understanding and controlling the stereochemistry of active pharmaceutical ingredients has become a fundamental aspect of drug discovery, development, and regulation.
Theoretical and Clinical Foundations
Theoretical Advantages of Single-Enantiomer Drugs
The pursuit of single-enantiomer drugs is driven by several compelling theoretical benefits that promise optimized pharmacotherapy:
- Enhanced Target Specificity: A pure, active enantiomer may interact more selectively with the intended biological target, potentially minimizing off-target interactions and reducing side effects [93].
- Reduced Therapeutic Dose: If the distomer is inactive or antagonistic, its elimination allows for a lower dose of the active moiety to achieve the same therapeutic effect, potentially enhancing patient safety and compliance [93].
- Simplified Pharmacokinetics: Single-enantiomer drugs can exhibit less inter-individual variability in absorption, distribution, metabolism, and excretion (ADME), leading to more predictable clinical dosing [93].
- Predictable Metabolic Profiles: Avoiding complex, stereoselective metabolism can simplify drug-drug interaction profiles and make clearance pathways more straightforward [93].
The Clinical Reality: A Case-by-Case Basis
Despite the strong theoretical framework, clinical evidence demonstrates that the superiority of a single enantiomer over its racemic counterpart is not guaranteed and must be empirically validated for each drug [93]. Regulatory agencies like the U.S. Food and Drug Administration (FDA) require manufacturers to demonstrate clinical superiority through comparative trials; improvements in pharmacokinetics or manufacturing control alone are insufficient without corresponding clinical benefits [93] [3]. A 2025 review by Agranat et al. strongly argues that "racemic drugs are not necessarily less efficacious and less safe than their single-enantiomer components," highlighting that racemates remain viable and sometimes preferable candidates [94] [95].
Table 1: Clinical Outcomes of Selected Single-Enantiomer Drugs vs. Their Racemic Counterparts
| Drug (Single Enantiomer vs. Racemate) | Clinical Finding | Conclusion on Advantage |
|---|---|---|
| Esomeprazole vs. Omeprazole | Improved acid control and healing rates, particularly at standard doses [93]. | Clinically meaningful advantage |
| Levocetirizine vs. Cetirizine | Similar efficacy with slightly fewer sedative effects [93]. | Modest advantage in side-effect profile |
| Dexmethylphenidate vs. Methylphenidate | Enabled similar efficacy at half the dose [93]. | Dosing advantage, similar clinical outcome |
| Escitalopram vs. Citalopram | Some studies suggest faster onset and greater symptom relief for depression [93]. | Clinically significant in some populations |
| S-Ibuprofen vs. Racemic Ibuprofen | No significant clinical difference; R-enantiomer undergoes metabolic inversion to the active S-form in vivo [93]. | No significant clinical difference |
Experimental and Methodological Approaches
Key Research Reagent Solutions
The development and analysis of chiral drugs require specialized reagents and techniques to separate, quantify, and characterize individual enantiomers.
Table 2: Essential Research Reagents and Materials for Chiral Drug Development
| Reagent / Material | Function in Chiral Drug Research |
|---|---|
| Chiral Resolving Agents | Chiral acids or bases used in classical racemic resolution to form diastereomeric salts that can be separated by crystallization [96]. |
| Chiral HPLC Columns | Stationary phases containing chiral selectors for the analytical or preparative separation of enantiomers via chromatography [96]. |
| Enantioselective Catalysts | Homogeneous or heterogeneous catalysts (including enzymes) designed to promote asymmetric synthesis, producing an excess of one enantiomer [96]. |
| Chiral Derivatizing Agents | Achiral reagents that react with enantiomers to form diastereomers, which can then be separated using standard achiral chromatographic methods [3]. |
Detailed Methodologies for Chiral Separation and Analysis
Classical Racemic Resolution
This traditional method involves treating a racemic mixture with a single enantiomer of a chiral resolving agent.
- Protocol:
- Reaction: The racemic drug (e.g., a free base) is reacted with an equimolar amount of a chiral resolving agent (e.g., a chiral acid) in a suitable solvent (e.g., ethanol) under heating to ensure complete salt formation.
- Crystallization: The solution is cooled slowly to facilitate the selective crystallization of the less soluble diastereomeric salt.
- Filtration and Washing: The crystals are collected via vacuum filtration and washed with a cold solvent to remove adhering mother liquor.
- Liberation: The pure enantiomer is liberated from the salt by treating the crystals with a base or acid, followed by extraction into an organic solvent and evaporation.
- Recycling: The mother liquor can be processed to isolate the opposite enantiomer or racemized for repeated resolution [96].
This method, while potentially wasting up to half of the material, is valued for being rapidly developed, cost-effective, and yielding high chemical and optical purity [96].
Chiral Preparative High-Performance Liquid Chromatography (HPLC)
Chiral preparative HPLC is a powerful technique for obtaining pure enantiomers, especially during preclinical development.
- Protocol:
- Column Selection: A preparative-scale HPLC column with a chiral stationary phase (e.g., cyclodextrin, macrocyclic glycopeptide) is selected based on analytical-scale screening.
- Mobile Phase Optimization: The mobile phase composition (e.g., hexane/isopropanol or aqueous buffers) is optimized to achieve baseline separation of enantiomers while maintaining solubility.
- Sample Loading: The racemic compound is dissolved in a suitable solvent and injected onto the column.
- Fraction Collection: The eluate is monitored (e.g., by UV), and fractions containing the individual enantiomers are collected separately.
- Concentration: The fractions are concentrated under reduced pressure to yield the pure enantiomers [96].
This method is highly effective for quickly supplying multi-gram quantities of pure enantiomers for initial activity and toxicology assays, though it is often too expensive for large-scale manufacturing [96].
In Vivo Enantiomer Interconversion Assessment
A critical experiment in chiral drug development is to determine whether enantiomerization occurs in vivo, which can undermine the purpose of developing a single-enantiomer drug.
- Protocol:
- Dosing: Administer the pure single enantiomer (e.g., the R-enantiomer) to animal models or human subjects via the intended clinical route.
- Serial Blood Sampling: Collect blood samples at predetermined time points post-administration (e.g., 0.25, 0.5, 1, 2, 4, 8, 12, 24 hours).
- Chiral Bioanalysis: Analyze plasma samples using a validated stereospecific bioanalytical method (e.g., chiral LC-MS/MS) that can quantify each enantiomer independently.
- Pharmacokinetic Analysis: Calculate pharmacokinetic parameters (AUC, C~max~, T~max~, t~1/2~) for both the administered enantiomer and its mirror image that appears in the plasma.
- Data Interpretation: The presence of the opposite enantiomer in plasma confirms in vivo interconversion. The extent of interconversion is quantified by the ratio of AUCs for the formed enantiomer versus the administered one [95] [93].
This methodology was key to understanding drugs like ibuprofen, where the inactive R-enantiomer undergoes metabolic inversion to the active S-form, justifying the continued use of the racemate [93].
Decision Framework and Development Workflow
The decision to develop a chiral drug as a single enantiomer or a racemate is multifaceted, involving scientific, clinical, and commercial considerations. The following workflow outlines a rational decision-making process for drug development professionals, integrating key experimental findings.
Diagram 1: Chiral Drug Development Workflow
This workflow emphasizes that development should "proceed forward with the racemate until and unless some relevant differentiated property is identified" [95]. Critical decision nodes include:
- In Vivo Interconversion: If rapid interconversion occurs in vivo, as with ibuprofen, developing the racemate is often more pragmatically and economically justified [95] [93].
- Clinical Superiority: For a single enantiomer to be favored, it must demonstrate statistically and clinically meaningful improvements in efficacy, safety, or pharmacokinetics in head-to-head trials against the racemate [93].
- Chiral Switch Evaluation: Even if a single enantiomer is developed subsequently ("chiral switch"), the racemic form may remain a therapeutically valid and cost-effective option, raising ethical and economic considerations about patient access and cost [93] [97].
The choice between developing a single-enantiomer drug and its racemic mixture is a complex technical and strategic decision that must be grounded in robust experimental and clinical evidence. While the theoretical advantages of single enantiomers are compelling, case studies like esomeprazole and escitalopram, which showed clear benefits, stand in contrast to others like ibuprofen, where the racemate remains the clinical standard due to in vivo interconversion [93]. The prevailing sentiment in the field is that racemic drugs are not intrinsically less efficacious or safe than their single-enantiomer components [94] [95]. A rational, data-driven approach that involves thorough stereochemical pharmacokinetic and pharmacodynamic evaluation early in development is paramount. As the field advances, the integration of pharmacogenomics into chiral drug development may enable more personalized, enantiospecific therapies, ensuring that the selected mirror image truly translates to optimal healing for the patient.
Overcoming Challenges in Stereochemical Analysis and Separation
Addressing Conformational Flexibility in Stereochemical Assignments
The determination of molecular configuration and conformation represents a cornerstone of organic chemistry research, with profound implications for understanding biological activity, material properties, and chemical reactivity. Stereochemical assignments provide the essential three-dimensional context needed to predict molecular behavior. However, a significant challenge arises from conformational flexibility—the ability of molecules to adopt multiple low-energy three-dimensional structures through rotation around single bonds. This flexibility complicates stereochemical analysis because experimental data often represents a dynamic average across all accessible conformers rather than a single, static structure [98]. For researchers in drug development, where the bioactive conformation determines pharmacological activity, accurately addressing this flexibility is paramount. Flexible molecules exist as ensembles of interconverting conformers at room temperature, and traditional ensemble analytical methods can only reflect the average contribution from all these states, potentially obscuring critical structure-activity relationships [99]. This technical guide examines contemporary approaches for resolving stereochemistry in flexible molecular systems, providing detailed methodologies for researchers navigating this complex analytical landscape.
Computational Approaches for Conformational Sampling and Analysis
Computational methods provide powerful tools for exploring the conformational landscape of flexible molecules. These techniques generate ensembles of low-energy structures that help interpret experimental data and identify biologically relevant conformations.
Advanced Sampling Algorithms
The single-coordinate-driving (SCD) method represents an early approach to traversing low-energy regions of conformational space. While effective for small to medium-sized flexible molecules, SCD can fail for more rigid yet conformationally interesting systems, such as RNA trimers. This limitation is overcome by coupling SCD with simulated annealing (SCD-SA), which enhances the search capability and eliminates sampling problems [100]. For more comprehensive sampling, the CREST program (Conformer-Rotamer Ensemble Sampling Tool) utilizes extensive sampling based on the semi-empirical extended tight-binding method (GFN2-xTB) combined with metadynamics to efficiently explore the low-energy conformational space. CREST identifies and groups rotamers—conformers identical except for atom re-indexing—and assigns probabilities based on Boltzmann distributions [101].
Benchmarking Computational Methods
The accuracy of computational methods for predicting conformational energies varies significantly. A recent benchmark study evaluated common methods against the highly accurate DLPNO-CCSD(T) coupled cluster theory, with the following results for mean error in conformational energy predictions [102]:
Table 1: Performance of Computational Methods for Conformational Energies
| Method Category | Specific Method | Mean Error (kcal mol⁻¹) |
|---|---|---|
| Quantum Mechanical | MP2 | 0.35 |
| Quantum Mechanical | B3LYP | 0.69 |
| Quantum Mechanical | HF (Hartree-Fock) | 0.81-1.00 |
| Force Fields | MMFF94 | 1.30 |
| Force Fields | MM3-00 | 1.28 |
| Force Fields | MM3-96 | 1.40 |
| Force Fields | MMX | 1.77 |
| Force Fields | MM+ | 2.01 |
| Force Fields | MM4 | 2.05 |
| Force Fields | DREIDING | 3.63 |
| Force Fields | UFF | 3.77 |
These benchmarks are crucial for selecting appropriate methods in drug design workflows, where accurate conformational energies ensure proper ranking of bioactive conformations.
Large-Scale Conformational Databases
The GEOM dataset (Geometric Ensemble Of Molecules) addresses a critical gap in computational chemistry by providing 37 million molecular conformations for over 450,000 molecules. This extensive collection includes conformers for 133,000 species from QM9 and 317,000 species with experimental data related to biophysics, physiology, and physical chemistry [101]. Such datasets enable machine learning models to predict properties from conformer ensembles and facilitate the development of generative models that sample 3D conformations more efficiently than traditional methods.
NMR Spectroscopy for Configurational Analysis in Flexible Systems
Nuclear Magnetic Resonance (NMR) spectroscopy remains the most powerful experimental technique for stereochemical assignment in solution, where molecules sample their conformational landscape. Specialized NMR approaches have been developed to address the challenges posed by molecular flexibility.
J-Based Configuration Analysis (JBCA)
For flexible molecules with multiple chiral centers, particularly acyclic and macrocyclic systems, traditional NMR parameters like vicinal proton-proton coupling constants (³JH,H) and nuclear Overhauser effects (NOEs) may be insufficient for complete configurational assignment. The J-based configuration analysis (JBCA) strategy, introduced by Murata and colleagues, utilizes two- and three-bond heteronuclear coupling constants (²JH,C and ³JH,C) to enhance relative configuration assignments [103].
The JBCA method exploits the Karplus-like dependency of ³JH,C values on dihedral angles. Although ²JH,C values are typically independent of dihedral angles, this dependence becomes relevant when the α-carbon is bonded to an electronegative atom (O, N, or halogen), where the ²JH,C value depends on the dihedral angle between the proton and the electronegative atom on the α-carbon [103]. The JBCA workflow involves:
- Measuring heteronuclear coupling constants for key structural fragments
- Relating coupling constants to dihedral angles using modified Karplus equations
- Identifying predominant rotamers in flexible systems
- Integrating with NOE/ROE data to resolve ambiguous cases
This approach is particularly valuable for 1,2- and 1,3-related stereogenic centers in type-I polyketide-derived natural products, which often contain multiple oxygenated chiral centers along flexible carbon chains [103].
Integrating NOE and RDC Data
Quantitative NOE measurements and residual dipolar couplings (RDCs) provide complementary information for conformational analysis. NOEs yield information through space, helping to establish through-space proximity between atoms, while RDCs provide orientation information relative to an external alignment tensor. However, both techniques present challenges in flexible molecules:
- NOE artifacts: Strongly coupled spin systems can produce artefacts in quantitative NOE spectra, which can be identified and corrected through computational simulations [98]
- Dynamic averaging: Both NOEs and RDCs represent ensemble averages across all populated conformations, requiring careful interpretation
- Complementary application: Using both NOEs and RDCs provides a more robust determination of configuration and conformation, as demonstrated in studies of strychnine and α-methylene-γ-butyrolactone [98]
Diagram 1: NMR Workflow for Flexible Molecules. This workflow integrates multiple NMR parameters to resolve stereochemistry in flexible systems.
Chemical and Single-Molecule Approaches
Chemical Derivatization Strategies
Chemical methods remain essential for stereochemical assignment, particularly for establishing absolute configuration. The Mosher's method using MTPA (methoxy(trifluoromethyl)phenylacetyl) reagents is widely employed for determining absolute configuration of secondary alcohols and amines. However, for flexible molecules with multiple chiral centers, this approach requires careful execution:
- Multiple derivatization: Generating Mosher esters at different sites can help triangulate absolute configuration
- Conformational effects: The flexibility of the MTPA ester chain can lead to complex conformational behavior, potentially complicating analysis
- Complementary reagents: Using different chiral derivatizing agents can provide validation through consistent results
In type-I polyketide-derived natural products with multiple hydroxy groups, improper derivatization or elimination of alcohol groups can lead to misinterpretations of absolute configuration [103].
Single-Molecule Techniques
Ensemble averaging presents a fundamental limitation for studying flexible molecules at room temperature. Single-molecule methods overcome this limitation by probing individual molecules, enabling the identification and characterization of conformational isomers that are obscured in ensemble measurements [99].
A groundbreaking study demonstrated this approach with cyclohexane, a classic example of ring inversion between chair conformers. Using single-molecule conductance measurements, researchers could:
- Quantitatively identify chair isomers of cyclohexane at room temperature
- Stabilize and characterize the twist-boat intermediate through confinement effects when electrodes attach to molecules
- Measure properties of individual conformers rather than ensemble averages
This methodology provides a promising strategy for studying conformational isomers of flexible molecules in contexts where they play important roles, including molecular biology, medicine, and supramolecular chemistry [99].
Table 2: Research Reagent Solutions for Stereochemical Analysis
| Reagent/Resource | Function/Application | Key Considerations |
|---|---|---|
| CREST Software | Conformer-rotamer ensemble sampling using GFN2-xTB | Provides Boltzmann-weighted conformer probabilities; more accurate than force fields [101] |
| MTPA (Mosher's) Reagent | Determination of absolute configuration via ¹H NMR | Requires careful interpretation for molecules with multiple chiral centers [103] |
| GEOM Dataset | Benchmarking conformer ensembles for machine learning | Contains 37 million conformations with energy annotations [101] |
| Chiral Solvating Agents | NMR-based discrimination of enantiomers | Complementary to Mosher's method for absolute configuration |
| Alignment Media (for RDCs) | Measuring residual dipolar couplings | Provides orientation constraints for conformational analysis [98] |
Integrated Workflow for Complex Systems: Type-I Polyketide Case Study
Type-I polyketide synthase-derived natural products exemplify the challenges in stereochemical analysis of flexible molecules. These compounds often contain multiple chiral centers along flexible carbon chains, requiring integrated approaches for complete configurational assignment [103]. The following workflow has proven effective:
- Initial structure elucidation using standard 1D and 2D NMR techniques
- Comprehensive conformational sampling using CREST or similar tools
- JBCA analysis of key fragments to establish relative configuration
- Chemical derivatization at strategic positions for absolute configuration
- Computational validation using quantum mechanical methods (B3LYP or MP2)
- Comparison with experimental chiroptical properties (ECD, VCD) when available
This integrated approach successfully addressed the stereochemical challenges in compounds like gibbosol, a super carbon-chained polyketide with multiple hydroxy chiral centers along its extended carbon chain [103].
Diagram 2: Integrated Approach to Stereochemical Assignment. Combining computational, spectroscopic, and chemical methods provides the most robust solution for flexible molecules.
Addressing conformational flexibility in stereochemical assignments requires moving beyond traditional static structural models to dynamic ensemble-based approaches. The integration of computational sampling with advanced NMR techniques, chemical derivatization, and emerging single-molecule methods provides a robust framework for tackling this challenge. For drug development professionals, these methodologies enable more accurate prediction of bioactive conformations and structure-activity relationships, ultimately supporting the design of more effective therapeutic agents. As computational power increases and experimental techniques continue to advance, the field moves closer to comprehensive characterization of molecular flexibility and its functional consequences.
Resolving Complex Chiral Systems with Multiple Stereocenters
Chirality, the geometric property of a rigid object (or spatial arrangement of points or atoms) of being non-superimposable on its mirror image, is a fundamental consideration in pharmaceutical research and development [104]. The vast majority of therapeutic small molecules contain one or more stereogenic centers—atoms with permutations of substituents that generate stereoisomers [104]. For molecules with multiple stereocenters, the complexity increases exponentially; a compound with n stereocenters can have up to 2^n stereoisomers. These include enantiomers (mirror-image pairs) and diastereomers (non-mirror-image stereoisomers) [54] [105] [80]. Diastereomers possess different physical and chemical properties, while enantiomers exhibit identical scalar properties but differ in their interaction with chiral environments, such as biological systems [54] [106].
The critical importance of stereochemistry in drug development stems from Nature's intrinsic homochirality. Enantiomers frequently exhibit profound differences in their pharmacological activity, metabolism, and toxicity [106]. Well-known cases like thalidomide underscore the necessity for enantiopure drugs. Consequently, developing efficient methods for the stereodivergent synthesis and resolution of complex chiral systems is paramount for producing safe and effective pharmaceuticals [107] [108]. This guide examines classical and emerging strategies for resolving such complex systems, with a focus on technical execution and contemporary advances.
Foundational Concepts: Stereochemistry and Isomerism
A clear understanding of isomerism is essential for discussing chiral resolution. Isomers are compounds with the same molecular formula but different structures, broadly categorized into constitutional isomers and stereoisomers [105] [80].
- Constitutional Isomers: These have the same molecular formula but different atomic connectivity. Subtypes include skeletal, functional, and positional isomers [80].
- Stereoisomers: These share the same atomic connectivity but differ in the spatial arrangement of their atoms. The two primary subcategories are enantiomers and diastereomers [105] [80].
Enantiomers are pairs of stereoisomers that are non-superimposable mirror images of each other. A molecule with a single stereogenic center exists as a pair of enantiomers. Diastereomers are stereoisomers that are not mirror images. This includes geometric isomers (e.g., cis-trans isomers across a double bond) and molecules with multiple stereocenters where some, but not all, configurations are inverted [54] [80]. Meso compounds are a special class of molecules that contain stereogenic centers but are optically inactive due to an internal plane of symmetry [80].
The following diagram illustrates the hierarchical relationships between these different types of isomers.
The effectiveness of any chiral resolution technique hinges on creating a temporary diastereomeric relationship between the enantiomer of interest and a chiral environment (e.g., a resolving agent, a chiral stationary phase, or polarized light), which allows for their differentiation and separation [106] [104].
Classical and Contemporary Resolution Methodologies
Crystallization-Based Techniques
Crystallization remains a cornerstone technique for industrial-scale chiral resolution.
- Diastereomeric Salt Crystallization: This is the most common crystallization method. A racemic mixture of a chiral acid (or base) is reacted with an enantiopure chiral base (or acid), the resolving agent, to form a pair of diastereomeric salts. These salts have different solubilities, allowing one to be selectively crystallized and filtered out. The pure enantiomer is then regenerated by acid/base treatment [109]. A modern application is in the synthesis of the drug duloxetine, where (S)-mandelic acid is used to resolve a racemic alcohol intermediate [109].
- Spontaneous and Preferential Crystallization: Approximately 5-10% of racemic compounds crystallize as a conglomerate, a physical mixture of crystals each containing only one enantiomer. These can be separated manually, as Louis Pasteur first demonstrated. Preferential crystallization (or "resolution by entrainment") involves seeding a supersaturated solution of a conglomerate with a crystal of the desired enantiomer, inducing the selective crystallization of that enantiomer from the racemate [109].
Chromatographic Methods
Chromatography is the predominant analytical and preparative method for chiral separation in research laboratories.
- Principle: All chromatographic methods (HPLC, GC, SFC) separate enantiomers by leveraging a chiral selector within the stationary phase. The enantiomers form transient diastereomeric complexes with the selector, and the strength of their non-covalent interactions (e.g., hydrogen bonding, π-π stacking, steric effects) determines their retention time, enabling separation [104].
- Common Techniques:
- Chiral HPLC (High-Performance Liquid Chromatography): The most widely used method, offering high efficiency, robustness, and applicability to a broad range of compounds.
- Chiral SFC (Supercritical Fluid Chromatography): Uses supercritical CO₂ as the mobile phase, offering faster separations and reduced solvent consumption compared to HPLC, making it more environmentally friendly and cost-effective for preparative-scale work [106].
- Chiral CE (Capillary Electrophoresis): A high-efficiency microfluidic technique where enantiomers are separated in a capillary filled with a run buffer containing a chiral selector, under the influence of a high-voltage electric field [106].
Kinetic and Dynamic Kinetic Resolution
These catalytic strategies resolve racemates by exploiting differences in the reaction rates of enantiomers with a chiral catalyst.
- Kinetic Resolution (KR): A chiral catalyst preferentially reacts with one enantiomer from a racemic mixture, converting it to a different product at a faster rate than the other enantiomer. The major limitation is the 50% theoretical yield for the desired enantiomer, as the other enantiomer remains unreacted [108].
- Dynamic Kinetic Resolution (DKR): This powerful strategy overcomes the yield limitation of KR by combining it with an in situ racemization of the starting material. As the fast-reacting enantiomer is consumed, the slow-reacting enantiomer is racemized, allowing for up to 100% theoretical yield of a single enantiomerically pure product [108]. The success of DKR relies on the racemization rate (krac) being much faster than the reaction rate of the asymmetric transformation (ka and k_b) [108].
Table 1: Key Chiral Resolution Techniques - A Comparative Overview
| Technique | Principle | Key Applications | Advantages | Limitations |
|---|---|---|---|---|
| Diastereomeric Salt Crystallization | Formation and separation of diastereomeric salts via selective crystallization [109]. | Industrial-scale production of enantiopure acids/bases (e.g., Duloxetine synthesis) [109]. | Scalable, cost-effective for large volumes. | Requires suitable functional groups; empirical screening of resolving agents. |
| Chiral Chromatography | Transient diastereomeric complex formation with a chiral stationary phase [104]. | Analytical & preparative separation of a wide range of chiral compounds. | High efficiency, broad applicability, high predictability. | High solvent consumption (HPLC); requires specialized, expensive columns. |
| Kinetic Resolution (KR) | Preferential reaction of one enantiomer with a chiral catalyst [108]. | Synthesis of enantiomerically enriched alcohols, epoxides, etc. | High enantioselectivity possible. | Maximum 50% yield for the desired enantiomer. |
| Dynamic Kinetic Resolution (DKR) | KR coupled with in situ racemization of the substrate [108]. | Synthesis of chiral alcohols, amines from racemic ketones, etc. [108]. | Bypasses 50% yield limit; high atom economy. | Requires compatible racemization and reaction conditions. |
Emerging Strategies for Complex Systems
Stereodivergent Synthesis and Dynamic Radical Recombination
Traditional resolution separates pre-formed racemates, but asymmetric synthesis builds the desired stereochemistry from achiral starting materials. A recent groundbreaking strategy for constructing complex chiral systems is stereodivergent synthesis, which allows access to all possible stereoisomers of a molecule from the same starting materials.
A prime example is the dynamic radical recombination (DRR) strategy for constructing spirocycles with 1,3-non-adjacent stereocenters [107]. This cobalt-hydride-catalyzed olefin hydroalkylation uses ligand control to dictate diastereoselectivity.
- Mechanism: Different chiral bisoxazoline ligands (L1 and L2) induce distinct mechanistic pathways. With ligand L1, the spirocyclic alkylcobalt(III) intermediate undergoes direct reductive elimination to afford the cis-spirocyclic product. Ligand L2 triggers a DRR pathway, where the intermediate undergoes reversible C–Co bond homolysis, followed by configurational inversion and radical recombination before reductive elimination, selectively yielding the trans-spirocyclic product [107].
- Significance: This represents a pioneering example of diastereodivergent and enantioselective construction of non-adjacent stereocenters via metal-hydride catalysis, providing a general route to rigid chiral spirocycles relevant for drug discovery [107].
The experimental workflow for this stereodivergent synthesis is outlined below.
Unconventional and External Stimuli-Triggered Methods
Several innovative approaches leveraging external stimuli are emerging as complementary tools for chiral resolution.
- Electro-Assisted Methods: These techniques use electric fields to enhance enantioselective separation. An electric field influences the movement of charged chiral species, allowing for selective migration of one enantiomer when combined with a chiral selector (e.g., in a capillary or on an electrode surface). They operate under mild conditions, reduce solvent needs, and offer high precision, making them attractive for sensitive pharmaceutical compounds [106] [110]. Wireless electro-assisted resolution using bipolar electrochemistry further eliminates the need for direct electrical connections, offering greater flexibility [110].
- The Chiral-Induced Spin Selectivity (CISS) Effect: This groundbreaking phenomenon enables enantiomer separation based on their interaction with electron spins. The CISS effect allows for additive-free enantioselective crystallization. By applying a magnetic field, the spin-selective interactions between chiral molecules and electron spins can influence the crystallization process to favor one enantiomer, achieving high purities without chemical resolving agents [106] [110].
- Advanced Chiral Selectors: New materials are being developed as chiral selectors for adsorption and membrane filtration. Homochiral Metal-Organic Frameworks (MOFs) and Covalent Organic Frameworks (COFs) act as solid-state host cavities with specific chirality, selectively adsorbing one enantiomer from a racemic mixture [106]. Chiral porous graphene membranes and functionalized microchannel membranes have also shown promise for enantioselective filtration [106].
Table 2: Performance Metrics of Selected Advanced Resolution Techniques
| Technique / System | Analyte / Substrate | Key Performance Metric | Reported Outcome |
|---|---|---|---|
| Dynamic Radical Recombination (DRR) [107] | Spirocyclic olefins (e.g., 1a) | Diastereomeric Ratio (dr), Enantiomeric Ratio (er) | >20:1 dr, >99:1 er |
| Ru-catalyzed DKR/Asymmetric Hydrogenation [108] | α-Amino-β-ketoester (4) | Enantiomeric Excess (ee), Diastereomeric Ratio (dr) | 99% ee, 85:15 dr |
| Chiral MOF Adsorption [106] | (S)-1-(1-naphthyl) ethanol | Enantiomeric Excess (ee) | 99% ee |
| Pillar[6]arene Functionalized Membrane [106] | (R)-phenylglycinol | Enantiomeric Excess (ee) | >90% ee |
Experimental Protocols
This protocol describes the ligand-controlled stereodivergent synthesis of spirocyclic compounds with 1,3-non-adjacent stereocenters.
Reaction Setup:
- In an inert atmosphere glovebox, add CoCl₂ (15 mol%) and chiral bisoxazoline ligand (L1 or L2, 18 mol%) to an oven-dried vial.
- Add anhydrous solvent (MTBE for L1, DME for L2, 0.5 mL).
- Stir the mixture for 10 minutes to pre-form the catalytic complex.
- Add spirocyclic olefin 1a (0.1 mmol, 1.0 equiv), alkyl iodide 2a (0.2 mmol, 2.0 equiv), K₃PO₄·H₂O (2.5 equiv), and DMMS (dimethoxymethylsilane, 3.0 equiv).
Reaction Execution:
- Cap the vial and remove it from the glovebox.
- Stir the reaction mixture at 0°C for 96 hours.
Work-up and Analysis:
- Quench the reaction by adding a saturated aqueous NH₄Cl solution.
- Extract the aqueous layer three times with ethyl acetate.
- Combine the organic extracts, dry over anhydrous Na₂SO₄, filter, and concentrate under reduced pressure.
- Purify the crude residue by flash column chromatography on silica gel.
- Analyze the enantiomeric excess (ee) by chiral HPLC and the diastereomeric ratio (dr) by ¹H NMR spectroscopy of the purified product (3a).
This protocol outlines a ruthenium-catalyzed dynamic kinetic resolution for the synthesis of enantiomerically pure anti β-hydroxy α-amino esters.
Reaction Setup:
- Charge a pipes-in-series flow reactor with a solution of (R,R)-Ts-DENEB-ligated ruthenium catalyst (cat.1, 0.5 mol%) in the appropriate solvent.
- Prepare a separate solution of racemic α-amino-β-ketoester 4 and the HCOOH/Et₃N azeotrope (5:2) as the hydrogen donor.
Reaction Execution:
- Pump both solutions through the flow reactor system, maintaining precise control over residence time, temperature, and pressure.
Work-up and Analysis:
- Collect the reactor effluent and concentrate under reduced pressure.
- Purify the product 5 via recrystallization.
- Determine the enantiomeric excess (ee) by chiral HPLC or GC and the diastereomeric ratio (dr) by ¹H NMR spectroscopy. The reported outcome is 99% ee and 85:15 dr, which can be improved to >99:1 dr after recrystallization [108].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Chiral Resolution
| Reagent / Material | Function / Application | Example / Notes |
|---|---|---|
| Chiral Bisoxazoline (BOX) Ligands | Control enantioselectivity and diastereoselectivity in metal-catalyzed reactions. | L1 and L2 ligands used in Co-catalyzed stereodivergent hydroalkylation to access cis- or trans-products, respectively [107]. |
| Chiral Ruthenabicyclic Complexes | Catalysts for asymmetric hydrogenation and transfer hydrogenation under DKR conditions. | cat.2 used for large-scale (147 kg) synthesis of lactone precursor 7 with >98:2 er [108]. |
| Chiral Resolving Agents | Form diastereomeric salts for classical resolution via crystallization. | (S)-Mandelic Acid: Used in the industrial resolution of Duloxetine intermediate [109]. 1-Phenylethylamine: A common base for resolving chiral acids [109]. |
| Chiral Metal-Organic Frameworks (MOFs) | Solid adsorbents for enantioselective separation via host-guest interactions. | Carboxylated-decorated MOF with 3D helical chirality for resolving (S)-1-(1-naphthyl) ethanol (99% ee) [106]. |
| Chiral Stationary Phases (CSPs) | The active component in chiral chromatography columns (HPLC, SFC, GC). | Various chemistries (e.g., polysaccharide-based, macrocyclic glycopeptide, Pirkle-type) coated on silica particles for analytical and preparative separations [106] [104]. |
| Silane Reducing Agents | Hydride source in metal-hydride-catalyzed reductions and hydroalkylations. | DMMS (Dimethoxymethylsilane): Used in the Co-catalyzed hydroalkylation protocol to generate the cobalt-hydride active species [107]. |
The field of chiral resolution is continuously evolving to meet the demands of modern drug discovery, which involves increasingly complex molecular architectures. While classical techniques like diastereomeric salt crystallization and chiral chromatography remain indispensable workhorses, the future lies in the development and integration of more efficient and sustainable strategies.
The advent of stereodivergent synthesis, as exemplified by the dynamic radical recombination approach, provides unprecedented control over the three-dimensional structure of molecules, allowing chemists to access the entire stereochemical space of a target [107]. Dynamic kinetic resolution continues to be refined, overcoming fundamental yield limitations and being applied to new substrate classes, including axially chiral compounds and N-heterocycles [108]. Meanwhile, unconventional methods leveraging electrical fields (Electro-assisted) and electron spin (CISS effect) offer promising, often more sustainable, pathways for enantiopurification that can complement or potentially replace traditional methods [106] [110].
Looking forward, the integration of machine learning is poised to revolutionize the field. Predictive models are being developed to forecast the outcomes of chiral resolutions, such as the optimal chiral stationary phase for a given molecule or the sign of its optical rotation, which could dramatically reduce the time and cost associated with empirical method screening [111]. As these computational and experimental tools converge, the resolution of complex chiral systems will become more predictive, efficient, and scalable, accelerating the delivery of new, stereochemically pure therapeutics to the market.
Separation Difficulties in Complex Biological Matrices
The precise separation and identification of individual molecules within complex biological matrices represents one of the most significant challenges in modern analytical chemistry, particularly in pharmaceutical development and metabolomics research. This challenge becomes exponentially more difficult when dealing with stereoisomers—molecules with identical atomic connectivity but differing three-dimensional spatial arrangements. Within the broader context of stereochemistry and isomerism research in organic molecules, the biological matrix introduces formidable complications that can obscure critical analytical data [112]. Biological samples such as plasma, serum, urine, and tissue homogenates contain innumerable interfering substances including salts, lipids, peptides, metabolites, and carbohydrates that co-elute with target analytes, potentially masking the presence of key stereoisomers and leading to inaccurate quantification [113].
The significance of overcoming these separation difficulties extends far beyond analytical curiosity. Stereoisomers frequently exhibit dramatically different biological activities, pharmacokinetic profiles, and toxicological effects in pharmaceutical contexts. For instance, the cis and trans isomers of phospholipids incorporating trans fatty acids have been linked to markedly different physiological effects and disease risks, including cardiovascular disease and metabolic disorders [114]. Similarly, the pharmacological activity of drug molecules can vary tremendously between enantiomers. Thus, the ability to successfully separate and characterize isomeric compounds within complex biological environments is not merely an analytical exercise but a fundamental requirement for drug safety and efficacy assessment.
Core Challenges in Separation Science
Matrix Effects in Biological Samples
Matrix effects represent perhaps the most pervasive challenge in the analysis of complex biological samples, particularly when utilizing highly sensitive detection methods like mass spectrometry. These effects occur when co-eluting compounds from the biological matrix alter the ionization efficiency of target analytes, leading to either ion suppression or enhancement that compromises accurate quantification [113]. In liquid chromatography-mass spectrometry (LC-MS), the most commonly observed matrix effect is ion suppression, which can significantly reduce method sensitivity and reliability [115].
The composition of biological matrices varies considerably between sample types, with each presenting unique interference profiles. Plasma and serum contain phospholipids, proteins, and lipids that are particularly problematic for maintaining analytical integrity [113]. These endogenous components compete with target analytes for ionization during the electrospray process, with phospholipids being especially notorious for causing significant ion suppression in LC-MS analyses [113]. The mechanisms behind matrix effects include competition for available charges in the liquid phase, increased droplet surface tension reducing analyte transfer to the gas phase, and co-precipitation of analytes with non-volatile matrix components [113] [115].
Structural Complexity of Isomeric Compounds
The separation of isomeric compounds presents unique challenges that extend beyond general matrix effects. Isomers possess identical mass-to-charge ratios, making them indistinguishable by mass spectrometry alone without effective chromatographic separation prior to detection [112]. The structural nuances that differentiate isomers—including regioisomerism, double bond position, stereochemistry, and enantiomerism—require highly selective separation techniques capable of resolving these subtle differences [112].
Table 1: Classification of Lipid Isomers and Analytical Challenges
| Isomer Type | Structural Difference | Analytical Challenge | Common Techniques |
|---|---|---|---|
| sn-Position isomers | Acyl chain esterification position on glycerol backbone | Inadvertent assignment without sufficient structural data | Specialized fragmentation techniques, extra analysis |
| Double bond position isomers | Location of carbon-carbon double bonds | MS alone cannot differentiate without pre-separation | Ozonolysis, ion mobility spectrometry, GC |
| Cis/Trans stereoisomers | Spatial orientation around double bonds | Nearly identical chemical properties | Silver ion HPLC, careful RP-LC optimization |
| Enantiomers | Chirality around asymmetric centers | Identical in most separation systems | Chiral chromatography, derivatization |
The challenges are particularly pronounced for lipid isomers, where subtle structural differences can significantly impact biological function. As noted in research on lipid structural characterization, "typical lipidomics analyses are achieved via MS, which as a sole technology is incapable of determining double bond geometry" [112]. This limitation necessitates sophisticated orthogonal approaches that combine separation science with advanced detection methodologies.
Advanced Separation Methodologies
Chromatographic Techniques for Isomer Separation
Reversed-phase liquid chromatography (RP-LC) has emerged as a powerful tool for separating isomeric compounds, particularly when optimized for specific separation challenges. The fundamental principle governing RP-LC separation of cis-trans isomers lies in their differential hydrophobicity and molecular geometry. Trans isomers, being more linear, behave chromatographically more like saturated straight-chain molecules and typically elute later than their bent cis equivalents due to increased interaction with the hydrophobic stationary phase [114].
The application of RP-LC to separate cis-trans isomers of phosphatidylglycerol and phosphatidylcholine with two 18:1 side chains demonstrates this principle effectively. When analyzing mitochondrial and serum samples from rats fed diets enriched in either trans or cis 18:1 fatty acids, researchers achieved baseline separation of these isomers, enabling quantitative assessment of their relative abundance in biological systems [114]. This approach allowed them to directly relate the cis:trans isomer ratio of phosphatidylcholine (18:1/18:1) to dietary composition, highlighting the biological relevance of such separations.
Silver ion chromatography represents another powerful technique specifically valuable for separating unsaturated compounds based on double bond geometry. This method leverages the selective complex formation between silver ions and π-electrons of double bonds, with cis isomers typically forming more stable complexes than trans isomers due to favorable orbital overlap [112]. Despite its utility, silver ion chromatography presents implementation challenges for mass spectrometric detection due to potential source contamination and signal suppression.
Innovative Sample Preparation Techniques
Effective sample preparation is crucial for mitigating matrix effects and enhancing the separation of isomeric compounds in complex biological matrices. Modern approaches have moved beyond traditional techniques like liquid-liquid extraction and protein precipitation, which often prove inadequate for removing problematic phospholipids and other interfering compounds [116].
Table 2: On-line Sample Processing Techniques for Biological Matrices
| Technique | Principle | Advantages | Applications |
|---|---|---|---|
| On-line Solid Phase Extraction (SPE) | Selective adsorption/elution on functionalized sorbents | Automated, high recovery, reduced matrix effects | Plasma, serum, tissue homogenates |
| Solid Phase Micro-Extraction (SPME) | Equilibrium extraction using coated fibers | Minimal solvent, integration of extraction/concentration | Environmental analysis, bioanalysis |
| Restricted Access Media (RAM) | Size exclusion of macromolecules with selective binding | Simultaneous protein removal and analyte enrichment | Direct injection of biological fluids |
| Turbulent Flow Chromatography (TFC) | High flow rates for size-based separation | Direct injection, efficient removal of proteins | High-throughput bioanalysis |
On-line solid phase extraction (SPE) coupled with LC-MS has demonstrated particular utility for analyzing complex biological matrices. This approach integrates sample purification, concentration, and introduction into the analytical system in a single automated workflow. For example, monolithic-phase based on-line SPE combined with HPLC-MS/MS has been successfully applied to determine drugs like amprenavir and atazanavir in human plasma, demonstrating reproducible and reliable quantitative data over hundreds of plasma injections while satisfying Good Laboratory Practice requirements for bioanalysis [116].
Experimental Protocols
Protocol for Cis-Trans Phospholipid Isomer Separation
The following detailed methodology has been adapted from established protocols for separating and characterizing cis-trans phospholipid isomers in biological matrices [114]:
Sample Preparation:
- Lipid Extraction: Employ a modified Bligh and Dyer method, substituting dichloromethane for chloroform. For mitochondrial samples (containing 1 mg protein), first dissolve in 40 μL DMSO and disrupt membranes by sonication.
- Internal Standard Addition: Add 30 μL of appropriate internal standard to each 30 μL sample (mitochondria or serum).
- Extraction Procedure: Add 190 μL methanol followed by 380 μL dichloromethane, vortex for 20 seconds, then add 120 μL water to induce phase separation. Vortex for 10 seconds and allow equilibration at room temperature for 10 minutes.
- Centrifugation: Centrifuge at 8000 g for 10 minutes at 10°C. Collect the organic (lower) phase and evaporate under nitrogen stream.
- Reconstitution: Reconstitute dried lipids in 100 μL acetonitrile/isopropanol/water (65:30:5, v/v/v) for LC-MS analysis.
LC-MS Conditions:
- Column: Reversed-phase C18 column (e.g., 2.1 × 100 mm, 1.8 μm)
- Mobile Phase A: Acetonitrile/water (60:40, v/v) with 10 mM ammonium formate
- Mobile Phase B: Acetonitrile/isopropanol (10:90, v/v) with 10 mM ammonium formate
- Gradient Program: Linear gradient from 30% B to 100% B over 20 minutes, hold at 100% B for 5 minutes
- Flow Rate: 0.4 mL/min
- Column Temperature: 55°C
- Injection Volume: 5 μL
- Ionization Mode: Positive and negative electrospray ionization
- Detection: High-resolution mass spectrometry with data-dependent MS/MS fragmentation
This method successfully achieves baseline separation of cis-trans isomers of phosphatidylglycerol and phosphatidylcholine standards, with the trans isomer eluting approximately 0.5 minutes after the cis isomer due to its increased hydrophobicity [114].
Protocol for Assessment and Mitigation of Matrix Effects
Post-extraction Addition Method for Matrix Effect Assessment:
- Prepare analyte standards in neat solvent at multiple concentration levels.
- Prepare equivalent standards in blank biological matrix extracts from at least six different sources.
- Compare the slope of the calibration curves between neat standards and matrix-matched standards.
- Calculate matrix effect (ME) using the formula: ME (%) = (slopematrix/slopeneat) × 100%
- A value of 100% indicates no matrix effects, while values <100% indicate suppression and >100% indicate enhancement.
Strategies for Matrix Effect Mitigation:
- Sample Dilution: When sensitivity permits, dilute samples to reduce concentration of interfering compounds.
- Enhanced Sample Cleanup: Incorporate selective SPE sorbents targeting phospholipids, such as those with zirconia-coated surfaces.
- Chromatographic Optimization: Adjust gradient conditions to shift analyte retention away from regions of high matrix interference.
- Internal Standardization: Use stable isotope-labeled internal standards (SIL-IS) that co-elute with analytes and experience equivalent matrix effects.
- Standard Addition Method: For endogenous analytes, employ standard addition by spiking known amounts of analyte into sample aliquots to account for matrix effects [115].
Figure 1: Experimental Workflow for Isomer Analysis in Biological Matrices
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Separation of Isomers in Biological Matrices
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Ammonium Formate | Mobile phase additive for LC-MS | Volatile salt that enhances ionization and chromatographic performance |
| Stable Isotope-Labeled Internal Standards | Compensation for matrix effects | Must be added prior to extraction to account for recovery variations |
| Monolithic SPE Sorbents | On-line sample cleanup | High capacity, low backpressure, suitable for direct plasma injection |
| Zirconia-Coated SPE Plates | Selective phospholipid removal | Critical for reducing major source of ion suppression in bioanalysis |
| Silver Ion Chromatography Materials | Cis-trans isomer separation | Specialized technique requiring dedicated instrumentation |
| Chiral Derivatization Reagents | Enantiomer resolution | Creates diastereomers with different chromatographic properties |
| HILIC Stationary Phases | Polar isomer separation | Complementary to reversed-phase for different isomer classes |
Visualization of Separation Mechanisms
Figure 2: Analytical Challenges and Solutions for Isomer Separation
The separation of stereoisomers in complex biological matrices remains a formidable challenge at the intersection of analytical chemistry, pharmaceutical science, and metabolomics. The interplay between matrix effects and the subtle structural differences between isomers demands sophisticated analytical approaches that combine optimized sample preparation, high-resolution separation techniques, and advanced detection methodologies. As research continues to reveal the critical biological significance of isomeric forms in drug action, metabolic regulation, and disease pathogenesis, the development of increasingly refined separation strategies will remain essential for advancing human health and therapeutic interventions.
The integration of orthogonal separation mechanisms—such as combining reversed-phase chromatography with ion mobility spectrometry or silver ion chromatography—represents a promising direction for overcoming current limitations. Furthermore, the continued development of automated on-line sample processing techniques will enhance analytical throughput and reliability while minimizing manual intervention and potential introduction of artifacts. As these methodologies evolve, they will undoubtedly expand our understanding of stereochemistry in biological systems and enable more precise characterization of isomeric compounds in complex matrices.
{# The Challenge of Metabolite Interference in LC-MS/MS Analysis of Isomers}
Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) is a cornerstone of targeted metabolomics, prized for its excellent quantitative capabilities and straightforward metabolite annotation [117]. However, the phenomenon of metabolite interference, where one metabolite produces a detectable signal in the mass transition of another, poses a significant threat to data accuracy [118]. This challenge is particularly acute for isomeric and isobaric metabolites, which share identical or nearly identical mass-to-charge ratios [118]. Within the broader context of stereochemistry and isomerism research, where the biological activity of a molecule is profoundly dependent on its three-dimensional structure [119], inaccurate identification or quantification can lead to flawed biochemical interpretations. This technical guide delves into the mechanisms of metabolite interference, provides evidence of its prevalence, and outlines detailed experimental protocols for its identification and mitigation, ensuring the reliability of targeted metabolomics data in drug development and related fields.
In organic chemistry and drug discovery, the biological activity of a molecule is often dictated by its stereochemistry [119]. Enantiomers, a type of stereoisomer, can exhibit vastly different pharmacological properties, uptake, and metabolism [119]. Liquid chromatography-tandem mass spectrometry (LC-MS/MS), particularly in Multiple Reaction Monitoring (MRM) mode, is a workhorse for quantifying specific metabolites in complex biological matrices. Its specificity arises from combining a metabolite's chromatographic retention time (RT) with its precursor ion (Q1) and a characteristic product ion (Q3) [118].
However, the unit mass resolution of triple-quadrupole (QQQ) instruments and the occurrence of in-source fragmentation can lead to metabolite interference [118]. This is especially problematic for isomers, which may not be fully separated by chromatography and can generate identical or similar fragment ions. Consequently, a signal attributed to a target "anchor" metabolite may originate from an "interfering" isomer, leading to misannotation and inaccurate quantification [118]. This directly compromises research aimed at understanding the distinct roles of stereoisomers in biological systems [119].
Mechanisms and Types of Metabolite Interference
Metabolite interference in LC-MS/MS can be classified into several types based on the underlying mechanism. A systematic analysis using metabolite standards has revealed that approximately 75% of metabolites generate a measurable signal in at least one other metabolite's MRM setting [118]. The following are the primary categories of interference.
- Isomeric Interference: This occurs when the anchor and interfering metabolites are true isomers, sharing the same molecular formula and, consequently, the same precursor ion (Q1). If their fragmentation patterns are similar, they may also share a common product ion (Q3), leading to identical MRM transitions [118]. Chromatographic separation becomes the only line of defense.
- Isobaric Interference: Isobars are molecules with different molecular formulas but the same nominal mass (e.g., differing in fine structure like
CH4vs.O). A QQQ mass spectrometer operating at unit resolution cannot distinguish between them in Q1, potentially leading to cross-talk in MRM channels [118]. - In-Source Fragmentation Interference: A metabolite prone to in-source decay can fragment before reaching the first quadrupole (Q1). The resulting in-source fragment ion, which is characteristic of the interfering metabolite, can then be selected by Q1 for the anchor metabolite's transition and subsequently fragmented in Q2, producing a signal that is misinterpreted as coming from the anchor metabolite [118].
The diagram below illustrates the analytical workflow and key points where these interferences can occur.
Figure 1: LC-MS/MS Workflow and Interference Points. In-source fragmentation and inadequate separation in Q1/Q3 are primary sources of interference.
Experimental Protocol for Identifying Metabolite Interference
A robust, multi-step experimental protocol is essential for systematically identifying and characterizing metabolite interference.
Materials and Reagent Solutions
Table 1: Key Research Reagents and Materials for Interference Analysis
| Item | Function / Description | Example / Specification |
|---|---|---|
| Metabolite Standards | Pure chemical compounds used to establish retention times, MRM parameters, and identify interfering pairs. | Purchased from commercial vendors (e.g., Selleck Inc.); prepared in DMSO or water at concentrations of 10 mM or 2 mM [118]. |
| LC-MS/MS System | Platform for separation and detection. | Triple quadrupole mass spectrometer (e.g., QTRAP 6500+); coupled with HPLC system [118]. |
| Chromatography Columns | Stationary phases for metabolite separation. | HILIC columns (e.g., iHILIC-(P) Classic, Waters XBridge Amide) for polar metabolite separation [118]. |
| Mobile Phases | Solvents for liquid chromatography. | Phase A: 20 mM ammonium acetate w/ 0.1% ammonium hydroxide; Phase B: Acetonitrile (ACN) [118]. |
| Data Processing Software | Converts and analyzes raw LC-MS data. | MSConvert (ProteoWizard) for file conversion; xcms R package for peak picking and alignment [118]. |
Step-by-Step Methodology
Step 1: Comprehensive Data Acquisition with Metabolite Standards
- Preparation: Group metabolite standards to avoid analytical interference during the initial screen [118].
- LC-MS/MS Analysis: Analyze each standard with the full set of MRM transitions in a single method. This generates a data matrix where the signal of every standard is checked across all MRM channels [118].
- Chromatography: Employ at least two distinct LC methods (e.g., using different HILIC columns) to assess the power of chromatographic resolution in eliminating interference [118].
Step 2: Data Processing and Peak Integration
- Convert raw data files (e.g.,
.wiff) to an open format likemzMLusingMSConvert[118]. - Use computational packages (e.g.,
xcmsin R) for peak detection, integration, and alignment across all samples and MRM transitions [118]. This creates a peak table with intensities for every standard in every MRM channel.
Step 3: Defining Interfering Metabolite Pairs (IntMPs) An Interfering Metabolite Pair (IntMP) is identified when an interfering metabolite produces a peak in the anchor metabolite's MRM transition that is chromatographically close (RT difference < 0.5 min) [118]. The following criteria are used to confirm interference:
- Spectral Evidence: The Q1 and Q3 m/z values of the anchor metabolite must be present in the MS2 spectrum of the interfering metabolite [118].
- Peak Shape Similarity: The cosine similarity between the interfering peak (in the anchor's MRM) and the true peak of the interfering metabolite (in its own MRM) should be >0.8 [118].
- Transition Ratio: The ratio of the interfering metabolite's intensity in the anchor's MRM over its intensity in its own MRM is calculated. A ratio ≥ 0.001 indicates significant interference [118].
Step 4: Assessing Interference in Biological Samples
- Annotate peaks in biological samples by matching them to expected RT and MRM transitions.
- Screen for potential interference using the pre-defined IntMP list from standards.
- For any suspect peak, check for the presence of the interfering metabolite in the sample by examining its unique MRM transition. Re-annotate the peak if the signal is determined to originate from the interfering metabolite based on transition ratio and peak shape similarity [118].
Quantitative Data on Metabolite Interference
Empirical data from a systematic study using 334 metabolite standards provides a sobering view of the prevalence and impact of this issue.
Table 2: Prevalence of Metabolite Interference from a Study of 334 Standards
| Interference Metric | Finding | Implication |
|---|---|---|
| Metabolites Causing Interference | ~75% of metabolites generated a signal in at least one other metabolite's MRM setting [118]. | Interference is the rule, not the exception, in complex targeted methods. |
| Resolution by Chromatography | Different LC methods resolved 65-85% of the interfering signals found among standards [118]. | Chromatographic optimization is a powerful but incomplete solution. |
| LC-Specific Interference Pairs | 88 pairs of metabolites showed interference that co-eluted (RT diff. < 0.5 min) in a specific LC method [118]. | Even with a good method, numerous specific interferences persist. |
| Mis-annotation in Biological Samples | ~10% of ~180 annotated metabolites in cell lysate and serum were mis-annotated or mis-quantified due to interference [118]. | The impact on real-world data is substantial and can invalidate biological conclusions. |
The following diagram summarizes the experimental and computational pipeline for identifying IntMPs.
Figure 2: Experimental Pipeline for Identifying Interfering Metabolite Pairs (IntMPs).
Strategies for Mitigation and Best Practices
To ensure data accuracy, researchers should adopt a proactive and multi-faceted approach to manage metabolite interference.
- Systematic Interference Screening: The most critical step is to perform a dedicated interference study using pure metabolite standards, as detailed in Section 3, to create a library of known IntMPs for your specific LC-MS method [118].
- Chromatographic Optimization: Since different LC methods resolve 65-85% of interferences, investing in method development is highly effective. Testing different columns (e.g., various HILIC chemistries, reversed-phase) and mobile phase gradients can achieve baseline separation of problematic isomeric pairs [118].
- Leveraging High-Resolution Mass Spectrometry (HRMS): While QQQ is excellent for quantification, orthogonal analysis using HRMS can distinguish isobars by their exact mass and provide confident metabolite identification, helping to validate findings from targeted assays [120] [117].
- Rigorous Data Interrogation: Manually inspect chromatographic peaks for shape, width, and retention time consistency. Be skeptical of peaks that are broad, asymmetric, or show a slight RT shift compared to the standard. For critical isomers, use unique and high-intensity product ions for quantification [118].
Metabolite interference is a pervasive and often overlooked challenge in LC-MS/MS targeted metabolomics, with the potential to mislead research conclusions, particularly in the nuanced field of stereochemistry where isomeric identity is critical [119]. Evidence indicates that approximately 10% of metabolites in a typical profiling assay may be mis-annotated or mis-quantified due to this phenomenon [118]. Acknowledging this problem is the first step toward robust analytical science. By integrating the described experimental protocols—including systematic interference screening with standards, chromatographic optimization, and rigorous data validation—researchers can significantly improve the accuracy of their metabolite measurements. This diligence is fundamental for generating reliable data that advances our understanding of the specific biological roles of isomers in drug development and disease mechanisms.
Optimizing Chromatographic Conditions for Epimer Separation
Stereochemistry represents a fundamental dimension in organic molecules research, where stereoisomers share identical molecular formulas and atomic connectivity but differ in the three-dimensional spatial arrangements of their atoms [121]. Among these, epimers are a critical class of stereoisomers that differ in configuration at only one stereogenic center, making their separation particularly challenging yet essential in fields like pharmaceutical development [122]. The significance of stereochemistry is profoundly evident in drug discovery, where the spatial arrangement of atoms can drastically influence a compound's pharmacological properties, binding affinity, metabolic stability, and toxicity [23]. A seminal example is methadone, where the (R)-enantiomer provides therapeutic opioid effects for pain relief, while the (S)-enantiomer binds to the hERG protein and can lead to severe cardiac side effects, including heart attacks [23]. This underscores the critical importance of developing robust chromatographic methods to separate and analyze epimers and other stereoisomers to ensure drug safety and efficacy.
The broader thesis of stereochemistry-aware research is gaining momentum, with recent investigations demonstrating that stereochemical editing can serve as an effective strategy for developing advanced materials, including cage-like energetic compounds where different diastereomers exhibit distinct densities, stability, and performance despite identical molecular formulas [86]. This expanding relevance across chemistry disciplines necessitates advanced separation technologies. Liquid chromatography mass spectrometry (LC-MS) has emerged as the gold standard for such challenging separations, owing to its ability to differentiate compounds with nearly identical structures and masses [122] [123]. This technical guide provides an in-depth examination of current methodologies, with a specific focus on optimizing chromatographic conditions for effective epimer separation within modern research contexts.
Fundamental Challenges in Epimer Separation
The primary challenge in separating epimers stems from their extreme structural similarity. With differences confined to a single stereocenter, their physicochemical properties are nearly identical, resulting in minimal differential interaction with most conventional chromatographic phases. This often manifests as poor resolution, co-elution, and inadequate baseline separation in analytical methods.
A significant complication in analysis arises from the fact that epimers share identical mass-to-charge ratios, making them indistinguishable by MS detection alone without effective prior chromatographic separation [122]. This limitation is particularly problematic in complex biological matrices where low analyte concentrations further complicate detection and quantification. Traditional solutions often relied on chiral stationary phases, which can be expensive, method-specific, and require extensive method development time.
The problem extends beyond analytical detection to preparative-scale purification, where the isolation of pure epimers is essential for biological activity testing, toxicological evaluation, and the development of enantiopure pharmaceuticals. The convergence of these challenges has driven research toward innovative mobile phase engineering as a more flexible and immediately applicable solution than stationary phase modifications alone.
Mobile Phase Engineering: A Practical Approach to Optimization
Recent research has demonstrated that strategic modification of mobile phase additives presents a powerful, practical approach to enhancing epimer separation without requiring specialized columns or extensive instrumentation changes.
The Ammonium Hydroxide Breakthrough
A landmark 2025 study systematically investigated ammonium hydroxide (AH) as a mobile phase additive for separating peptide epimers/isomers with D-amino acid modifications and isoaspartate modifications [122]. This research revealed several significant advantages over traditional acidic additives like formic acid (FA) and trifluoroacetic acid (TFA), as summarized in the table below.
Table 1: Performance Comparison of Mobile Phase Additives for Peptide Epimer/Isomer Separation
| Additive Type | Concentration | Base Peak Intensity (MS1) | Number of Resolved Epimers/Isomers | Retention Order | Typical Run Times |
|---|---|---|---|---|---|
| Ammonium Hydroxide (AH) | 0.1% | 1.06-6.56 fold increase vs. acidic additives | Higher across all tested sets | Changed for some isomer sets | Shorter in some cases |
| Formic Acid (FA) | 0.1% | Baseline | Lower than AH | Standard reference | Standard |
| FA + TFA | 0.1% + 0.05% | Lower than AH | Lower than AH | Standard reference | Longer than AH |
The study tested different sets of peptide epimers/isomers with varying chain lengths and acidic, basic, and neutral side chains [122]. Key findings revealed that AH not only improved ionization efficiency but also enhanced chromatographic resolution and in some instances reduced analysis time. Furthermore, the observed changes in retention order for some epimer sets when using AH instead of acidic additives indicate a fundamentally different separation mechanism, providing an additional tool for method development when purifying specific peptide isomers.
Mechanism of Action
The improved performance with ammonium hydroxide stems from multiple synergistic effects. Basic conditions alter the charge state distribution of peptides, which can enhance separation based on subtle differences in pKa values around the epimeric center [122]. The volatile nature of ammonium hydroxide makes it particularly compatible with MS detection, eliminating the ion suppression effects commonly associated with non-volatile additives or trifluoroacetic acid. Additionally, the basic environment potentially influences the three-dimensional conformation of peptides in solution, potentially magnifying slight structural differences between epimers and facilitating their chromatographic resolution.
Experimental Protocol: Implementing Ammonium Hydroxide Methodology
Reagent Preparation
- Ammonium Hydroxide Mobile Phase: Add 1.0 mL of high-purity ammonium hydroxide (28% NH₄OH) to 1 L of LC-MS grade water to achieve a 0.1% (v/v) concentration. Prepare both aqueous (Mobile Phase A) and organic (Mobile Phase B, typically acetonitrile) solutions with identical additive concentrations.
- Peptide Standards: Dissolve synthetic peptide epimers in a suitable solvent (e.g., water/acetonitrile mixture) to prepare stock solutions of 1 mg/mL. Further dilute with the initial mobile phase composition to achieve final injection concentrations appropriate for MS detection.
- System Conditioning: Before analysis, condition the LC system and column with the ammonium hydroxide mobile phase for at least 10-15 column volumes to ensure stable baseline and reproducible retention times.
Chromatographic Conditions
- Column: C18 stationary phase (e.g., 100 × 2.1 mm, 1.8-2.7 μm particle size)
- Mobile Phase: A: 0.1% ammonium hydroxide in water; B: 0.1% ammonium hydroxide in acetonitrile
- Gradient: Optimize based on peptide hydrophobicity (e.g., 5-50% B over 20-30 minutes)
- Flow Rate: 0.2-0.4 mL/min
- Temperature: 30-40°C
- Injection Volume: 1-5 μL
- Detection: MS with electrospray ionization (ESI) in positive or negative mode, optimized for the target analytes
Method Optimization Strategy
- Gradient Slope Adjustment: For difficult separations, implement a shallower gradient (e.g., 0.5-1% B per minute) to enhance resolution, potentially at the cost of longer run times.
- Temperature Effects: Evaluate retention and resolution at 30°, 40°, and 50°C, as temperature can significantly impact the kinetics of interaction with the stationary phase.
- Additive Concentration: While 0.1% is standard, test 0.05% and 0.15% ammonium hydroxide to fine-tune separation, noting that higher concentrations may increase MS background noise.
- Column Screening: If separation remains challenging, supplement the method with alternative stationary phases such as C8, phenyl-hexyl, or polar-embedded groups while maintaining ammonium hydroxide in the mobile phase.
Diagram 1: Epimer separation method development workflow. This flowchart outlines the systematic approach to developing and optimizing chromatographic methods for epimer separation using ammonium hydroxide-based mobile phases.
Advanced Techniques and Complementary Approaches
Two-Dimensional Liquid Chromatography (LC×LC)
For exceptionally complex samples containing multiple epimeric pairs, comprehensive two-dimensional liquid chromatography (LC×LC) provides significantly enhanced separation power [124]. Recent advancements include multi-2D LC×LC systems that employ a six-way valve to select between different separation mechanisms (e.g., HILIC or RP) in the second dimension based on elution time in the first dimension [124]. This approach is particularly valuable when epimers are part of complex mixtures with other isomeric compounds, as it provides an additional separation dimension that can resolve co-eluting species.
Ion Mobility Spectrometry (IMS) Integration
The integration of ion mobility spectrometry (IMS) with LC-MS systems adds yet another separation dimension based on the size, shape, and charge of gas-phase ions [125]. This technique is especially powerful for distinguishing isomeric and isobaric compounds, as it provides collision cross-section (CCS) values that serve as a unique physicochemical property for compound identification [125]. Building a CCS library for specific epimer classes, as demonstrated recently for pyrrolizidine alkaloids, creates a powerful identification tool that complements chromatographic retention time data [125].
Active Solvent Modulation
A significant technical challenge in LC×LC, particularly when combining different separation mechanisms, is the incompatibility between the eluent from the first dimension and the optimal starting conditions for the second dimension. Active solvent modulation (ASM) technology addresses this by adding a solvent (e.g., water for RP phase, acetonitrile for HILIC phase in the 2nd dimension) to reduce the elution strength before the second dimension separation, thereby improving peak focusing and resolution [124].
Diagram 2: Multi-dimensional separation strategy. This workflow illustrates how combining multiple separation dimensions (LC×LC) with ion mobility spectrometry and mass spectrometry creates a powerful platform for resolving and identifying complex mixtures of epimers and other isomers.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Research Reagent Solutions for Epimer Separation
| Reagent/Material | Function/Application | Key Considerations |
|---|---|---|
| Ammonium Hydroxide (NH₄OH) | Basic mobile phase additive for improved epimer separation and MS ionization | Use high-purity LC-MS grade; volatile and MS-compatible [122] |
| High-Purity Water & Acetonitrile | Mobile phase components for reversed-phase chromatography | LC-MS grade with low UV cutoff and minimal impurities |
| C18 Stationary Phases | Reversed-phase chromatography columns | Various particle sizes (1.8-5μm) and pore sizes for different peptides [122] |
| Alternative Phases (C8, Phenyl, HILIC) | Complementary separation mechanisms for challenging separations | Provides different selectivity when C18 fails [124] |
| Chiral Stationary Phases (CSPs) | Direct enantiomer/epimer separation without derivatization | Multiple classes available (polysaccharide, protein, cyclodextrin) [123] |
| Cation-Exchange SPE Cartridges | Sample clean-up and pre-concentration for complex matrices | Particularly useful for basic compounds like alkaloids [125] |
| Peptide Epimer Standards | Method development and validation | Commercially available or custom-synthesized [122] |
| Formic Acid & Trifluoroacetic Acid | Standard acidic mobile phase additives for comparison | Enable method comparison and selectivity studies [122] |
The optimization of chromatographic conditions for epimer separation represents a critical capability in modern stereochemistry research, with far-reaching implications for pharmaceutical development, materials science, and analytical chemistry. The strategic implementation of ammonium hydroxide as a mobile phase additive offers a significant advancement, providing improved resolution, enhanced MS detection sensitivity, and altered selectivity for challenging epimer separations [122]. When combined with advanced multidimensional separation platforms such as LC×LC and emerging detection technologies like ion mobility spectrometry, researchers now possess an unprecedented toolkit for tackling the most complex stereochemical challenges [124] [125]. As the field continues to recognize the profound biological and material implications of stereochemistry, these optimized separation methodologies will play an increasingly vital role in ensuring the efficacy, safety, and performance of molecular innovations across scientific disciplines.
The development of chiral drugs represents a critical crossroads in pharmaceutical research, where sophisticated science intersects with complex economic decision-making. Chirality, a fundamental property of nature, describes molecules that exist as non-superimposable mirror images, known as enantiomers [126]. Within the human body—itself a chiral environment built from L-amino acids and D-sugars—these mirror images can exhibit dramatically different biological activities [126]. The strategic choice between developing a drug as a racemic mixture (a 50:50 mixture of both enantiomers) or as a single enantiomer carries profound implications for therapeutic efficacy, safety profiles, development timelines, and ultimately, commercial viability.
Historically, approximately 90% of synthetic chiral drugs were marketed as racemates, primarily due to technical challenges and costs associated with separating enantiomers [126]. However, tragic lessons such as thalidomide—where one enantiomer provided therapeutic benefit while its mirror image caused severe birth defects—catalyzed a paradigm shift in regulatory thinking and industry practices [126]. This whitepaper examines the multifaceted economic considerations underlying this critical development decision, providing researchers and drug development professionals with a structured framework for optimizing stereochemical strategy within modern pharmaceutical development.
Scientific Foundation: Pharmacological Differences Between Enantiomers
Key Terminology and Biological Significance
The biological differentiation between enantiomers stems from their distinct three-dimensional interactions with chiral biological structures including enzymes, receptors, and transporters [126]. This understanding requires precise terminology:
- Eutomer: The enantiomer with the desired, greater physiological activity [126].
- Distomer: The opposing enantiomer, which may be inactive, less active, or contribute undesirable effects [126].
- Eudysmic Ratio: The quantitative ratio of activity between eutomer and distomer, providing a numerical basis for evaluating the potential benefit of developing a single enantiomer [126].
The "three-point attachment model" explains the molecular basis for enantioselectivity in drug-receptor interactions, where the eutomer achieves optimal alignment with complementary binding sites while the distomer experiences steric or electronic mismatch [126]. These differential interactions extend throughout the drug's pharmacokinetic profile—absorption, distribution, metabolism, and excretion (ADME)—as chiral environments throughout the body process each enantiomer differently [126].
Clinical Evidence: Variable Outcomes from Enantiomer Separation
Clinical experience with single-enantiomer drugs reveals a spectrum of outcomes that underscore the case-specific nature of development decisions:
Table: Clinical Outcomes of Selected Single-Enantiomer Drugs
| Drug (Single Enantiomer) | Racemic Predecessor | Clinical Outcome | Economic Impact |
|---|---|---|---|
| Esomeprazole [93] | Omeprazole | Improved acid control & healing rates | Significant market success |
| Escitalopram [93] [96] | Citalopram | Faster onset, greater efficacy in some patients | High sales despite premium price |
| Levocetirizine [93] | Cetirizine | Similar efficacy, slightly improved side effect profile | Moderate advantage |
| Dexmethylphenidate [93] | Methylphenidate | Similar efficacy at half the dose | Dosing advantage only |
| (S)-Ibuprofen [93] | Racemic ibuprofen | No significant clinical difference | Racemate remains standard |
The variable outcomes illustrated above demonstrate that scientific plausibility does not guarantee clinical success [93]. Each case must be evaluated based on hard clinical data and careful benefit-risk assessment rather than theoretical advantages alone.
Economic Analysis: Development Cost Considerations
Synthetic Route Economics
The production of enantiopure compounds presents significant cost considerations that impact development strategy. Three primary technical approaches exist, each with distinct economic profiles:
Table: Economic Comparison of Enantiomer Production Methods
| Production Method | Development Timeline | Cost Structure | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Asymmetric Synthesis [96] | Longer development cycle | Higher R&D investment, potentially lower production cost | Ideal for long-term manufacturing; "green" catalytic options emerging [127] | Technologically challenging; may not yield both enantiomers for testing |
| Chiral Chromatography [96] | Rapid implementation | High equipment and consumable costs | Excellent for preclinical stages; provides both enantiomers | Generally prohibitively expensive for large-scale manufacturing |
| Classical Racemic Resolution [96] | Quickly developed | Lower development costs; 50% material loss | Fast implementation; well-established; good chemical/optical purity | Inherent 50% yield limitation; waste disposal considerations |
The economic viability of these methods depends heavily on project phase. Asymmetric synthesis typically requires substantial upfront investment but may offer superior economics at commercial scale, particularly with advances in organocatalysis and biocatalysis that reduce reliance on precious metal catalysts [127]. Conversely, racemic resolution provides a cost-effective solution for early-stage development where speed to clinical trials is paramount [96].
Development Timeline and Regulatory Considerations
Regulatory requirements for stereochemically pure drugs introduce additional economic considerations. According to FDA guidelines, "manufacturers should develop quantitative assays for individual enantiomers in in vivo samples early in drug development" [96]. This necessitates:
- Earlier investment in chiral analytical methods
- Comprehensive pharmacokinetic studies of individual enantiomers
- Potential clinical trials comparing enantiopure and racemic forms
The stereochemistry market, projected to grow from $2.3 billion in 2024 to $5.4 billion by 2033 (12.5% CAGR), reflects increasing industry investment in chiral technologies [128]. This growth is driven by multiple factors including "growing demand for targeted drug delivery," "need for enantiomerically pure compounds," and "rising popularity of personalized medicine" [128].
The "Chiral Switch" Strategy: Patent Lifecycle Considerations
Strategic Patent Considerations
The "chiral switch"—developing a single enantiomer version of a racemic drug approaching patent expiration—represents a significant pharmaceutical strategy with dual scientific and commercial motivations [126]. When successfully executed, this approach can:
- Extend market exclusivity by 5-7 years
- Create a clinically superior product
- Potentially command premium pricing
However, this strategy carries substantial risk. Regulatory agencies require demonstration of clear clinical benefit rather than merely pharmacokinetic improvements [93]. Payers increasingly scrutinize cost-benefit ratios, with some implementing step therapy requirements mandating trials of generic racemates before funding single-enantiomer versions [93].
Ethical and Access Considerations
The chiral switch strategy raises important ethical questions, particularly when clinical advantages are marginal but price differentials are substantial [93]. Key considerations include:
- Affordability and access: Premium pricing may limit patient access, particularly in publicly-funded healthcare systems
- Informed consent: Patients should understand relative benefits when switching from racemic to single-enantiomer formulations
- Health equity: Prolonged market exclusivity may disproportionately affect underserved populations [93]
These factors underscore that the decision to pursue a chiral switch must balance commercial objectives with ethical responsibilities and public health impact.
Emerging Technologies and Future Directions
Advanced Analytical and Synthetic Technologies
Recent technological advances are reshaping the economic landscape of chiral drug development:
- AI and Machine Learning: Algorithms now predict enantioselective reaction outcomes and design chiral catalysts, reducing development timelines [127]
- High-Throughput Experimentation: Automated systems rapidly screen chiral catalysts and conditions, accelerating route optimization [127]
- Green Chemistry Approaches: Sustainable methodologies including organocatalysis and biocatalysis reduce environmental impact and manufacturing costs [127]
These innovations are making enantioselective synthesis increasingly economically viable across the development lifecycle.
The Scientist's Toolkit: Key Research Reagent Solutions
Table: Essential Research Reagents for Chiral Drug Development
| Reagent/Catalyst Type | Function | Application Examples |
|---|---|---|
| Organocatalysts [127] | Metal-free asymmetric catalysis | MacMillan/List catalysts for Diels-Alder, Michael additions |
| Enzyme Systems [127] | Biocatalytic resolution or asymmetric synthesis | Transaminases (e.g., sitagliptin synthesis); lipases for ester resolutions |
| Chiral Ligands [127] | Transition metal catalysis for asymmetric synthesis | BINAP, BOX ligands for hydrogenation, cyclopropanation |
| Chiral Solvating Agents [104] | NMR-based enantiopurity determination | Chiral lanthanide shift reagents for ee determination |
| Chiral Stationary Phases [104] | Chromatographic enantiomer separation | Polysaccharide, cyclodextrin, or Pirkle-type HPLC columns |
Strategic Decision Framework
Integrated Cost-Benefit Analysis
The 1995 seminal review "The cost benefit ratio of enantiomeric drugs" established that single-enantiomer development should only be pursued when "advantages of the eutomer in terms of efficacy and tolerability outweigh the associated increase in production and development costs" [129] [130]. This principle remains relevant today, though the economic calculus has evolved with technological advances. A modern decision framework must incorporate:
- Eudysmic ratio: Higher ratios (>100) strongly favor single-enantiomer development [126]
- Distomer safety profile: The presence of significant distomer toxicity favors enantiopure development
- Technical feasibility: Availability of economically viable synthetic routes
- Market considerations: Patent status, competitive landscape, and pricing potential
- Regulatory trajectory: Increasing emphasis on stereochemical characterization
Experimental Protocols for Early-Stage Evaluation
Protocol 1: Rapid Preclinical Enantiomer Profiling
- Compound Preparation: Utilize preparative chiral HPLC to obtain gram quantities of both enantiomers for initial screening [96]
- In Vitro Profiling: Conduct parallel pharmacological screening of both enantiomers and racemate across:
- Primary target binding/functional assays
- Selectivity panels (related targets)
- Early cytotoxicity assessments
- Pharmacokinetic Screening: Compare basic ADME properties including:
- Metabolic stability in liver microsomes
- Plasma protein binding
- Caco-2 permeability
Protocol 2: Synthetic Route Evaluation Matrix
- Parallel Route Development: Initiate concurrent evaluation of:
- Asymmetric synthesis approaches (catalytic, stoichiometric)
- Biocatalytic options (enzyme screening)
- Classical resolution methods [96]
- Economic Modeling: Project costs across development lifecycle and commercial scale
- IP Landscape Analysis: Evaluate freedom to operate and patent protection strategies
Decision Pathway Visualization
The following workflow diagrams the strategic decision process for enantiomer development:
The strategic choice between racemic mixture and single-enantiomer development remains a complex multidimensional decision requiring integrated analysis of scientific, clinical, technical, and commercial factors. While technological advances in asymmetric synthesis and analytical methods have made enantiopure development increasingly accessible, the economic case must be evaluated specifically for each drug candidate. The most successful development strategies will be those that balance potential therapeutic advantages with realistic assessment of development costs and market dynamics, ultimately aligning stereochemical strategy with broader development objectives and patient needs.
The future of chiral drug development will likely be shaped by precision medicine approaches, where patient subpopulations may derive differential benefit from specific stereoisomers [93]. Additionally, continuing advances in AI-driven catalyst design [127], continuous flow chiral synthesis [127], and green chemistry methodologies [127] promise to further improve the economic viability of enantioselective manufacturing across the pharmaceutical industry.
Regulatory Compliance in Stereoselective Assay Validation
Stereochemistry, the spatial arrangement of atoms within molecules, represents a fundamental consideration in modern drug discovery and development. The majority of biologically active compounds, including natural products and pharmaceutical substances, are chiral molecules that exist as non-superimposable mirror images known as enantiomers [119]. These enantiomeric pairs, while identical in most physical properties, can exhibit dramatically different biological activities within chiral environments such as biological systems. The profound implications of molecular handedness on drug action necessitate rigorous stereoselective assay validation to ensure therapeutic efficacy and patient safety.
The historical context of chirality in pharmaceuticals provides compelling evidence for the necessity of stereoselective analysis. The thalidomide tragedy of the 1960s serves as a stark reminder, where one enantiomer provided therapeutic relief for morning sickness while the other caused severe teratogenic effects [131]. Although subsequent research revealed that thalidomide enantiomers undergo rapid interconversion in vivo, this episode fundamentally shifted regulatory perspectives worldwide [132]. Similarly, contemporary research continues to demonstrate significant differences in enantiomeric activity, as evidenced by recent investigations into 3-Br-acivicin derivatives where only the (5S, αS) isomers displayed significant antiplasmodial activity against Plasmodium falciparum strains, while other stereoisomers showed dramatically reduced potency [119].
Within pharmaceutical development, stereoselective assay validation has evolved from a specialized consideration to a regulatory imperative. The Food and Drug Administration (FDA) and European Medicines Agency (EMA) now provide specific guidance on developing chiral drugs, emphasizing the need for stereospecific analytical methods when enantiomers demonstrate differences in pharmacokinetic or pharmacodynamic properties [132] [133]. This regulatory landscape demands robust, validated stereoselective assays capable of accurately quantifying individual enantiomers throughout drug development, manufacturing, and bioequivalence studies.
Regulatory Framework and Guidelines
Evolution of Regulatory Perspectives
The regulatory approach to chiral drugs has undergone significant transformation since the early 1980s, moving from acceptance of racemic mixtures to a clear preference for enantiomerically pure pharmaceuticals. This shift was catalyzed by seminal work such as E. J. Ariëns' 1984 paper, which asserted that ignoring stereoselectivity in drug action resulted in "highly sophisticated scientific nonsense" [132]. This growing recognition led to the publication of foundational regulatory documents, including the FDA's "Development of New Stereoisomeric Drugs" in 1992 and the EMA's "Investigation of Chiral Active Substances" in 1994 [132].
Current regulatory trends demonstrate a strong preference for single enantiomer drugs. Between 2013 and 2022, the EMA has not approved a single racemate since 2016, while the FDA averaged only one racemic drug approval per year during this period [132]. Those racemic drugs that do gain approval typically fall into specific categories: drugs previously marketed elsewhere for several decades, analogues of pre-existing drugs, or compounds where the undefined stereocenter does not play a role in therapeutic activity [132].
Key Regulatory Requirements
Regulatory agencies require comprehensive stereochemical assessment throughout the drug development process. Key requirements include:
- Stereospecific pharmacokinetic studies to understand the behavior of individual enantiomers, even in racemic mixtures [133]
- Assessment of chiral inversion potential in vivo, where one enantiomer may convert to its mirror image [133]
- Enantiomer-specific characterization of pharmacological activity, metabolism, and toxicity [132]
- Validated analytical methods capable of quantifying individual enantiomers in complex matrices [134]
For bioequivalence studies of chiral drugs, regulatory agencies recommend measuring individual enantiomers when they exhibit different pharmacokinetic or pharmacodynamic properties, particularly when one enantiomer is primarily responsible for therapeutic effects [133]. This emphasis on stereospecific analysis reflects recognition that conventional non-stereospecific methods may mask clinically significant differences between enantiomers.
Fundamental Validation Parameters for Stereoselective Assays
The validation of stereoselective analytical methods requires demonstration that established performance characteristics are suitable for their intended application. While general validation principles apply to stereoselective assays, specific considerations related to chiral separations necessitate specialized approaches.
Specificity and Selectivity
For stereoselective assays, specificity demonstrates the method's ability to unequivocally assess the analyte of interest in the presence of potential interferents, including the other enantiomer, metabolites, and matrix components. The separation of enantiomers requires creating a chiral environment through interaction with a chiral selector, forming transient diastereomeric complexes with different physicochemical properties that enable separation [135].
Selectivity verification must include:
- Resolution between enantiomeric pairs
- Separation from potential impurities and degradation products
- Discrimination from matrix components in biological samples
The selectivity parameter can be determined using approaches that adjust for interference bias, ensuring accurate quantification of individual enantiomers [134].
Accuracy, Precision, and Linearity
Accuracy reflects the closeness of agreement between the measured value and the true value of the enantiomer. For chiral assays, accuracy should be established for each enantiomer individually across the validation range, requiring enantiomerically pure reference standards [134].
Precision encompasses repeatability (intra-assay) and intermediate precision (inter-assay) and should be evaluated for each enantiomer at multiple concentration levels. Regulatory guidance emphasizes the importance of replicate recommendations to ensure reliable precision estimates [134].
Linearity demonstrates the ability of the method to obtain test results proportional to the concentration of each enantiomer within the specified range. While linear regression (R²) has been traditionally used, regulatory authorities have clarified thinking on the adequacy of this approach, noting that it should be complemented with other statistical measures [134].
Sensitivity Parameters
Sensitivity requirements for stereoselective assays include establishing the limit of detection (LOD) and limit of quantification (LOQ) for each enantiomer. Regulatory guidelines provide flexibility in determination approaches, including:
- Signal-to-noise ratio methods
- Spike and recovery approaches for determining LOD [134]
- Statistical methods based on the standard deviation of the response and the slope of the calibration curve
For bioanalytical applications, sensitivity must be sufficient to characterize the pharmacokinetic profile of each enantiomer throughout the anticipated concentration range.
Table 1: Key Validation Parameters for Stereoselective Assays
| Parameter | Requirements for Stereoselective Assays | Special Considerations |
|---|---|---|
| Specificity | Resolution of enantiomers ≥1.5; Separation from impurities and matrix | Chiral stationary phase selectivity; Mobile phase optimization |
| Accuracy | 85-115% recovery for each enantiomer | Requires enantiomerically pure standards; Matrix effects evaluation |
| Precision | RSD ≤15% for each enantiomer | Repeatability and intermediate precision for both enantiomers |
| Linearity | R² ≥0.990 across specified range | Evaluation for each enantiomer individually; Residual analysis |
| Range | Established from LOQ to upper quantification limit | Must cover expected concentrations of both enantiomers |
| LOD/LOQ | Signal-to-noise ≥3:1 for LOD; ≥10:1 for LOQ | Determined for each enantiomer; Sufficient for pharmacokinetic studies |
Experimental Design and Methodologies
Analytical Techniques for Chiral Separation
Multiple chromatographic techniques are available for stereoselective analysis, each with particular advantages for specific applications:
Chiral High-Performance Liquid Chromatography (HPLC) represents the most widely employed technology for enantiomer separation [135]. Chiral HPLC utilizes chiral stationary phases (CSPs) packed with chiral selectors that differentially interact with enantiomers through formation of transient diastereomeric complexes [136]. Common CSPs include polysaccharides, cyclodextrins, glycopeptide antibiotics, proteins, and Pirkle-type phases [135] [136]. Normal phase solvents are frequently used, though reversed-phase conditions are applicable with certain CSPs [136].
Supercritical Fluid Chromatography (SFC) has emerged as a powerful complementary technique, particularly for method screening and preparative separations. SFC typically uses carbon dioxide as the primary mobile phase component, offering advantages in separation efficiency and method speed [131]. Modern SFC systems enable parallel screening of multiple racemates, significantly increasing throughput [131].
Capillary Electrophoresis (CE) and Gas Chromatography (GC) provide alternative approaches for chiral separations. Chiral CE primarily employs cyclodextrins as chiral selectors added to the background electrolyte, while chiral GC typically uses cyclodextrin derivatives as stationary phases [135].
Method Development Workflow
The development of validated stereoselective methods follows a systematic workflow:
Chiral Method Development Workflow
Initial screening employs multiple CSPs with different mobile phase compositions to identify promising separation conditions. Advanced implementations use "chiral pooling" approaches where multiple analytes are injected from a single vial, with mass spectrometry detection enabling simultaneous evaluation of separation for multiple compounds [131].
Method optimization refines initial conditions to achieve baseline resolution (Rs ≥1.5) with acceptable analysis time. Critical parameters include:
- Mobile phase composition (organic modifier, additives, pH)
- Temperature
- Flow rate
- Gradient profile
Robustness testing evaluates method resilience to small, deliberate variations in operational parameters, establishing system suitability criteria for routine application.
Bioanalytical Method Validation for Chiral Drugs
For bioequivalence studies, stereospecific bioanalytical methods require validation according to regulatory guidelines [133] [134]. Key considerations include:
- Matrix effects evaluation for each enantiomer in the appropriate biological matrix
- Stability assessment of individual enantiomers under storage and processing conditions
- Extraction efficiency determination for each enantiomer
- Selectivity against endogenous matrix components and metabolites
Case studies demonstrate that non-stereospecific methods may fail to detect critical bioequivalence issues. For example, ibuprofen formulations appearing equivalent using non-stereospecific methods showed significant differences in the active S-enantiomer profile when analyzed with stereospecific assays [133].
Table 2: Comparative Techniques for Chiral Analysis
| Technique | Principle | Applications | Advantages |
|---|---|---|---|
| Chiral HPLC | Diastereomeric complex formation with CSP | Enantiomeric purity; Bioanalytical; Stability testing | Wide applicability; Robustness; Preparative capability |
| SFC | Chiral separation with CO₂-based mobile phase | Method screening; Purification; Polar compounds | Fast analysis; Green technology; High efficiency |
| Chiral CE | Differential migration with chiral selectors | Bioanalysis; Polar compounds; Ionizable analytes | High efficiency; Small sample volume; Aqueous compatibility |
| Chiral GC | Chiral stationary phases at elevated temperatures | Volatile compounds; Essential oils; Fragrances | High resolution; Sensitive detection; Thermally stable analytes |
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of stereoselective assays requires specialized materials and reagents carefully selected for each application:
Chiral Stationary Phases represent the core component of chiral chromatographic methods. Comprehensive chiral screening typically employs multiple CSP chemistries, with polysaccharide-based phases being the most widely applicable [131] [136]. These phases derive their selectivity from complex interaction mechanisms including hydrogen bonding, π-π interactions, dipole stacking, and steric effects [136]. Modern chiral columns are engineered for direct separation of diverse enantiomeric compounds including amines, alcohols, carboxylic acids, amino acids, and other biologically active substances [136].
Enantiomerically Pure Reference Standards are essential for method development, validation, and quantification. The availability of high-purity enantiomeric standards enables accurate determination of chromatographic elution order, calibration curve establishment, and method validation [135]. These standards must be thoroughly characterized for enantiomeric purity using orthogonal methods.
Mobile Phase Components require careful selection and purification. For normal-phase chiral HPLC, alcohol modifiers such as ethanol and isopropanol are commonly employed [136]. Mobile phase additives including acids, bases, or volatile salts may be necessary to optimize selectivity and peak shape. For bioanalytical applications, MS-compatible additives are preferred when using mass spectrometric detection.
Sample Preparation Materials must be evaluated for potential stereoselective effects. Solid-phase extraction cartridges, filtration devices, and solvents should be validated to ensure no racemization or preferential loss of either enantiomer occurs during sample processing.
Case Studies and Applications
Stereochemistry in Antimalarial Drug Development
Recent research on 3-Br-acivicin (3-BA) derivatives provides a compelling case study on the profound impact of stereochemistry on biological activity [119]. Investigation of all four stereoisomers of 3-BA revealed that only the (5S, αS) isomers displayed significant antiplasmodial activity against Plasmodium falciparum strains, with IC₅₀ values below 1 μM [119]. In contrast, isomers with (5R, αR) configuration showed moderate activity (IC₅₀ 1-10 μM), while (5S, αR) and (5R, αS) configurations resulted in poorly active compounds (IC₅₀ >20 μM) [119].
Table 3: Impact of Stereochemistry on Antimalarial Activity of 3-BA Derivatives
| Compound | Stereochemistry | P. falciparum D10 IC₅₀ (μM) | P. falciparum W2 IC₅₀ (μM) |
|---|---|---|---|
| 1a | (5S, αS) | 0.35 ± 0.08 | 0.34 ± 0.12 |
| 1b | (5R, αS) | 23.54 ± 0.31 | 24.75 ± 0.90 |
| 1c | (5S, αR) | 7.49 ± 1.48 | 8.47 ± 2.06 |
| 1d | (5R, αR) | 8.79 ± 1.12 | 10.18 ± 1.75 |
| 2a | (5S, αS) | 0.79 ± 0.21 | 0.88 ± 0.23 |
| 2b | (5R, αS) | 17.14 ± 5.91 | 17.18 ± 7.39 |
This research demonstrated that stereochemistry significantly influenced antimalarial activity across all compound subclasses, suggesting that stereoselective uptake mediated by the L-amino acid transport system might be responsible for the enhanced biological activity of the (5S, αS) isomers [119]. Molecular modeling further elucidated structural and stereochemical requirements for efficient interaction with Plasmodium falciparum glyceraldehyde 3-phosphate dehydrogenase (PfGAPDH), a key enzyme in the parasite's glycolytic pathway [119].
Bioequivalence Assessment of Chiral Drugs
Stereospecific bioequivalence studies have revealed significant limitations of conventional non-stereospecific methods. Case studies include:
Ibuprofen: A bioequivalence study using non-stereospecific methods concluded therapeutic equivalence, while stereospecific assays revealed significant differences in the pharmacokinetic profile of the active S-enantiomer [133]. This discrepancy has clinical implications since the S-enantiomer is primarily responsible for anti-inflammatory activity, while the R-enantiomer undergoes partial metabolic inversion to the active form [133].
Carvedilol: Stereospecific analysis identified variations in enantiomer activity that would have been masked by achiral methods [133]. This finding is particularly relevant as carvedilol enantiomers display different pharmacological activities—primarily β-blocking activity in one enantiomer and antioxidant properties in the other [133].
These case studies underscore the necessity of stereospecific bioanalytical methods for accurate bioequivalence assessment when enantiomers demonstrate different pharmacological activities or pharmacokinetic profiles.
Regulatory compliance in stereoselective assay validation represents an essential component of modern pharmaceutical development for chiral therapeutics. The profound influence of stereochemistry on drug efficacy, safety, and pharmacokinetics necessitates robust, validated analytical methods capable of accurately characterizing and quantifying individual enantiomers throughout the drug development lifecycle.
The current regulatory landscape demonstrates a clear preference for enantiomerically pure drugs, with requirements for comprehensive stereochemical assessment during development and approval. Successful navigation of this landscape requires implementation of validated stereoselective methods that address specificity, accuracy, precision, and sensitivity for each enantiomer individually. Advanced chromatographic techniques, particularly chiral HPLC and SFC, provide powerful tools for enantiomer separation and quantification when properly developed and validated.
As pharmaceutical research continues to explore increasingly complex chiral molecules, including atropisomers and other stereogenic units, the importance of stereoselective assay validation will only intensify. Adherence to regulatory guidelines, implementation of robust analytical methods, and comprehensive understanding of stereochemistry's implications on drug action will remain critical for delivering safe and effective chiral therapeutics to patients.
Validation Techniques and Comparative Analysis of Stereoisomers
Stereochemistry, the three-dimensional arrangement of atoms in molecules, serves as a fundamental determinant of biological activity, particularly in pharmaceutical research. The efficacy, safety, and metabolic fate of chiral therapeutic agents are profoundly influenced by their absolute configuration (AC) [23] [137]. Incorrect stereochemical assignments can lead to severe consequences, as exemplified by (S)-methadone, which is associated with cardiotoxic effects, in contrast to the therapeutic (R)-enantiomer used for pain relief [23]. This technical guide examines the synergistic application of X-ray crystallography, Circular Dichroism (CD), and Nuclear Magnetic Resonance (NMR) spectroscopy for the unequivocal determination of stereochemistry. Within the broader context of stereochemistry and isomerism research, this cross-validated approach is paramount for ensuring the structural integrity of organic molecules throughout the drug development pipeline, from discovery to quality control.
Theoretical Foundations of Stereochemistry
Principles of Chirality and Absolute Configuration
At its core, molecular chirality describes the geometric property of a molecule being non-superimposable on its mirror image. The Cahn-Ingold-Prelog (CIP) system provides the standardized nomenclature (R/S) for describing the absolute configuration of chiral centers [138]. The assignment process involves:
- Priority Assignment: Ranking the four substituents attached to the chiral center based on atomic number (higher atomic number receives higher priority).
- Orientation: Viewing the molecule along the bond from the chiral center to the lowest-priority substituent, which must be pointing away from the observer.
- Configuration Determination: Tracing a path from priority 1 to 2 to 3. A clockwise path denotes the R configuration (Latin: rectus), while a counterclockwise path denotes the S configuration (Latin: sinister) [138].
Table 1: Key Stereochemical Phenomena and Their Implications
| Phenomenon | Description | Structural/Biological Implication |
|---|---|---|
| Enantiomers | Non-superimposable mirror image molecules [23]. | Often exhibit different biological activities and optical activities [23]. |
| Diastereomers | Stereoisomers that are not mirror images [23]. | Exhibit different physical and chemical properties [23]. |
| Geometric Isomers (E/Z) | Arise from restricted rotation around a double bond [23]. | Can lead to distinct pharmacological profiles. |
| Conformational Lability | Ability of a molecule to adopt different shapes via bond rotation [137]. | Impacts biological activity and spectral analysis [137]. |
The Critical Need for Cross-Validation
While single-crystal X-ray diffraction is often considered the definitive method for absolute configuration determination, it is not infallible. Challenges can arise from poor crystal quality, the crystallization of an unrepresentative component of the bulk material, or the inherent difficulty of crystallizing certain compounds [137]. Furthermore, structures derived from crystallographic or NMR data are typically refined using stereochemical restraints derived from known small-molecule structures, which can mask local errors if the experimental data are weak [139]. Therefore, cross-validation using solution-based techniques like CD and NMR is not merely supplementary but is often essential for confirming stereochemical assignments with a high degree of confidence, ensuring that the structure determined in the solid state is representative of the bioactive form in solution [137].
Methodological Deep Dive: The Triad of Techniques
X-ray Crystallography
3.1.1 Principles and Workflow X-ray crystallography determines molecular structure by analyzing the diffraction pattern produced when X-rays interact with a crystalline sample. The positions and intensities of the diffracted beams are related to the electron density within the crystal via Bragg's Law (nλ = 2d sinθ) [140]. The process involves several key stages, as visualized below.
Diagram 1: X-ray crystallography workflow.
3.1.2 Advanced Applications and Limitations For stereochemical assignment, the Flack parameter is a crucial metric derived from crystallographic refinement, providing a statistical measure of the absolute structure [137]. Advanced techniques like ensemble refinement can model structural heterogeneity, creating multiple conformer models (ensembles) to better fit the diffraction data and provide insights into protein dynamics [141]. However, the accuracy of these ensembles in representing solution-state dynamics must be validated against solution-based techniques like Residual Dipolar Couplings (RDCs) from NMR [141]. The primary limitation of crystallography is the requirement for high-quality, diffracting crystals, which can be difficult or impossible to obtain for some molecules. Furthermore, the crystalline environment may not always reflect the conformation present in solution or at the biological target site [137].
Circular Dichroism (CD) Spectroscopy
3.2.1 Principles and Spectroscopic Ranges CD spectroscopy measures the difference in absorption of left- and right-handed circularly polarized light by chiral molecules. This differential absorption creates a spectrum that is highly sensitive to the absolute configuration, conformation, and electronic environment of the chromophore(s) involved [137]. Two primary forms are used:
- Electronic CD (ECD): Operates in the ultraviolet and visible range, probing electronic transitions. It is sensitive to chromophores and their immediate environment [137].
- Vibrational CD (VCD): Operates in the infrared range, probing vibrational transitions. It provides information about the entire molecular skeleton and is particularly powerful for determining AC in the absence of strong chromophores [137].
3.2.2 Experimental Protocol for Absolute Configuration Determination
- Sample Preparation:
- Prepare a solution of the chiral compound at an appropriate concentration (typically 0.1-1 mg/mL for ECD, higher for VCD) in a suitable solvent.
- Use high-purity solvents and accurately determine sample concentration, as errors here are a major source of inaccuracy (10-25% uncertainty is common) [142].
- Utilize cells with path lengths appropriate for the wavelength range (e.g., 0.1-1 mm for far-UV ECD).
- Data Acquisition:
- For ECD: Collect spectra typically from 180-260 nm (far-UV for secondary structure) or 250-350 nm (near-UV for aromatic/ligand effects). Perform measurements at controlled temperature.
- For VCD: Collect spectra in the mid-IR region (typically 1800-800 cm⁻¹). Signal averaging over many scans is necessary due to the small signals.
- Quantum Chemical Calculation:
- Generate a conformational ensemble by scanning the molecule's flexible torsion angles.
- Optimize the geometry of each low-energy conformer using density functional theory (DFT).
- Calculate the theoretical ECD or VCD spectra for each conformer.
- Boltzmann-weight and sum the spectra of all populated conformers to produce a final theoretical spectrum.
- Validation and Assignment:
- Compare the experimentally measured CD spectrum with the Boltzmann-weighted theoretical spectrum.
- A strong match between the experimental and calculated spectra for one enantiomer, and a mirror-image relationship for its opposite, allows for a confident assignment of AC [137].
3.2.3 Strengths, Limitations, and Validation Power CD spectroscopy excels at analyzing compounds in solution under near-physiological conditions. It is highly sensitive to conformational changes and, when combined with computational chemistry, provides a powerful tool for AC determination, especially when crystals for X-ray analysis are unavailable [137]. However, its accuracy can be compromised by several factors: scaling errors from concentration inaccuracies, deviations between the reference structure and the solution structure, and non-secondary structure contributions (e.g., from aromatic side chains or co-factors) [142]. The combined use of ECD and VCD can substantially increase the credibility of the assignment, as they provide complementary information [137].
Nuclear Magnetic Resonance (NMR) Spectroscopy
3.3.1 Principles and Key Parameters NMR spectroscopy exploits the magnetic properties of certain atomic nuclei to elucidate molecular structure and dynamics in solution. For stereochemistry, key parameters include:
- Scalar J-Couplings: Provide information on dihedral angles through the Karplus relationship, yielding relative configuration.
- Nuclear Overhauser Effect (NOE): Measures through-space dipolar interactions between nuclei, providing distance restraints crucial for determining relative stereochemistry and 3D structure [143].
- Residual Dipolar Couplings (RDCs): Measured for molecules weakly aligned in a magnetic field, RDCs provide long-range orientational restraints. They are highly precise reporters on the time-weighted average orientation of internuclear vectors and are exceptionally sensitive to dynamic processes and the quality of structural models [141].
3.3.2 Protocol for Structure Determination and Validation
- Data Collection:
- Acquire a standard set of multidimensional NMR experiments (e.g., COSY, TOCSY, NOESY, HSQC, HMBC) for resonance assignment.
- For RDCs, collect data in both isotropic and weakly aligned media (e.g., using phage or bicelles).
- Restraint Generation:
- Convert NOE cross-peak intensities into inter-proton distance restraints.
- Derive dihedral angle restraints from J-coupling constants.
- Extract RDC values by comparing coupling constants in isotropic and aligned media.
- Structure Calculation:
- Use distance geometry, simulated annealing, or molecular dynamics restrained by the experimental NOE, dihedral, and RDC data to generate an ensemble of structures (a "bundle") [143].
- Validation:
- Analyze the bundle for violations of experimental restraints and stereochemical quality. Key metrics include Clashscore, and the number of Ramachandran and sidechain dihedral angle outliers [143].
- Validate the NMR ensemble against independent data. For proteins, RDCs provide a superb cross-validation metric against X-ray structures or among themselves [141].
3.3.3 Comparison with X-ray Structures A fundamental question is whether differences between NMR and X-ray structures are methodological or reflect genuine environmental differences (solution vs. crystal). Evidence suggests that high-quality NMR structures, determined with ample experimental restraints (e.g., >10 NOE restraints per residue) and exhibiting good stereochemistry (low Clashscore, few Ramachandran outliers), possess hydrophobic core packing properties nearly identical to high-resolution X-ray structures [143]. This indicates that the proteins themselves are fundamentally the same in both environments, and earlier reported differences were likely methodological artifacts.
Integrated Cross-Validation Strategies
Decision Framework for Stereochemical Assignment
The choice of technique, or combination thereof, depends on the scientific question, sample properties, and available resources. The following diagram outlines a logical pathway for stereochemical assignment and validation.
Diagram 2: Decision pathway for stereochemical analysis.
Quantitative Comparison of Techniques
A holistic cross-validation strategy leverages the complementary strengths of each technique, as summarized in the table below.
Table 2: Comparative Analysis of Stereochemical Validation Techniques
| Parameter | X-ray Crystallography | Circular Dichroism (CD) | NMR Spectroscopy |
|---|---|---|---|
| Primary Information | 3D Electron Density Map | Differential Absorption of CPL | Through-Space & Through-Bond Interactions |
| Stereochemical Output | Absolute Configuration (via Flack parameter) [137] | Absolute Configuration & Conformation [137] | Relative Configuration & Conformational Ensemble |
| Sample State | Solid Crystal | Solution | Solution |
| Sample Requirement | Single Crystal (~nL volume) | ~0.1-1.0 mg (ECD/VCD) | ~1-10 mg (for small molecules) |
| Key Strengths | Atomic resolution; Definitive AC assignment [137] | High sensitivity to AC/conformation; Fast measurement [137] | Atomic-level detail in solution; Probes dynamics [141] |
| Key Limitations | Requires crystallization; Crystal packing effects [137] | Requires chromophore (ECD); Computational complexity [137] | Low sensitivity; Resonance overlap in large molecules |
| Key Validation Metrics | R-free factor, Ramachandran plot, Clashscore [139] | Fit between experimental and calculated spectra (RMSD) [142] | Restraint violations, Clashscore, RDC Q-factor [141] [143] |
Case Study: Integrating X-ray and RDCs for Protein Dynamics
For the SARS-CoV-2 main protease (Mpro), ensemble models derived from X-ray diffraction data using the ECHT method showed substantially improved agreement with solution NMR RDC data compared to a single, conventionally refined model, especially for dynamic protein regions [141]. Remarkably, a weighted ensemble of 350 independent Mpro X-ray structures provided slightly better cross-validated RDC agreement than any individual ensemble refinement, highlighting the power of combining data from multiple sources to average out uncertainties and model dynamic behavior more accurately [141]. This case exemplifies a successful cross-validation workflow where NMR RDCs serve as a sensitive benchmark for evaluating and improving dynamic models derived from crystallographic data.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Stereochemical Analysis
| Reagent/Material | Function and Application |
|---|---|
| Chiral Solvating Agent (CSA) | Engages in diastereomeric interactions with analyte enantiomers in NMR, causing chemical shift differences for signal discrimination. |
| Alignment Media (e.g., Pf1 Phage, Bicelles) | Induces weak molecular alignment in solution for the measurement of Residual Dipolar Couplings (RDCs) in NMR [141]. |
| High-Purity Spectroscopic Solvents | Essential for CD and NMR to avoid interfering absorbance bands or signal artifacts. |
| Crystallization Screening Kits | Contain diverse precipitant and condition solutions to empirically identify optimal parameters for growing diffraction-quality crystals. |
| Stereochemical Restraint Libraries (e.g., Engh & Huber) | Provide target values for bond lengths, angles, and other parameters during the refinement of X-ray and NMR structures [139]. |
The unambiguous assignment of stereochemistry is a non-negotiable requirement in modern organic and medicinal chemistry research. As demonstrated, no single technique is universally sufficient to address all challenges. X-ray crystallography can provide a definitive structural model but is constrained by the crystalline state. CD spectroscopy offers a potent solution-based method for determining absolute configuration but relies heavily on computational support. NMR spectroscopy delivers unparalleled insights into solution-state structure and dynamics, yet can typically only provide relative configuration without additional tools. A cross-validated approach, leveraging the synergistic strengths of two or more of these techniques, is the most robust strategy. This integrated methodology ensures that stereochemical assignments are accurate, reliable, and reflective of the molecular species' true nature, thereby de-risking drug discovery and solidifying the foundational science of isomerism.
Within the broader context of stereochemistry and isomerism research, the comparative pharmacokinetics of enantiomers presents a critical area of study with profound implications for drug discovery and development. Stereoisomers, particularly enantiomers—mirror images of a chiral molecule—often exhibit distinct biological activities despite sharing identical molecular formulas and atom connectivity [144]. This difference arises because biological systems, composed of chiral environments like proteins, enzymes, and receptors, can distinguish between these two forms, much as a left hand fits into a left-handed glove but not a right-handed one [145].
The investigation into the Absorption, Distribution, Metabolism, and Excretion (ADME) properties of individual enantiomers is therefore not merely an academic exercise but a fundamental requirement for developing safer and more effective therapeutics. The tragic case of thalidomide in the mid-20th century, where one enantiomer provided the desired sedative effect while the other caused severe teratogenic effects, starkly highlighted the pharmacological significance of chirality [145]. This review provides an in-depth technical examination of the ADME differences between enantiomers, framing them within the essential principles of stereochemistry and highlighting the advanced experimental and computational methodologies used to characterize them.
Fundamental Principles of Stereochemistry in Pharmacology
Key Definitions and Concepts
- Chirality: A geometric property of a molecule that lacks an internal plane of symmetry, making it non-superimposable on its mirror image. The carbon atom is a common chiral center when it has four different substituents.
- Enantiomers: A pair of stereoisomers that are mirror images of each other.
- Diastereomers: Stereoisomers that are not mirror images, often arising from molecules with multiple chiral centers.
- Eutomer and Distomer: In a pair of enantiomers, the eutomer is the form with the higher pharmacological activity, while the distomer is the form with lower activity [145].
- Eudismic Ratio: The quantitative ratio of the activities (e.g., EC50 or IC50) of the eutomer to the distomer. A high ratio indicates significant stereoselectivity at the target site, providing a strong rationale for developing a single-enantiomer drug [145].
- Racemate: A 50:50 mixture of two enantiomers, often referred to as a racemic mixture.
The Molecular Basis for Stereoselective Recognition
The primary reason enantiomers exhibit different pharmacokinetic and pharmacodynamic profiles lies in stereoselective recognition within the body. Biological macromolecules such as enzymes, transporters, receptors, and plasma proteins are themselves chiral. Their binding sites are three-dimensional and asymmetric, designed to accommodate specific molecular shapes. Consequently, one enantiomer may fit perfectly into a binding pocket, while its mirror image may fit poorly or interact with different amino acid residues altogether [145]. This differential binding is the root cause of disparities in ADME properties and ultimately, in therapeutic and toxicological outcomes.
Mechanisms of ADME Differences Between Enantiomers
Stereoselectivity can influence every stage of a drug's journey through the body. The following sections detail the mechanisms and consequences for each ADME process.
Absorption
Drug absorption, particularly via active transport processes, can be stereoselective. Passive diffusion across biological membranes is generally not stereoselective, as it depends largely on lipophilicity and molecular size, which are identical for enantiomers. However, when absorption involves chiral carrier-mediated transporters in the gastrointestinal tract, enantiomers may be absorbed at different rates or extents.
- Example: The absorption of L-amphetamine is more rapid and complete than that of D-amphetamine from the gut, due to differential affinity for chiral transporters [145].
Distribution
Once in the systemic circulation, a drug's distribution to its sites of action and elimination is influenced by plasma protein binding and tissue partitioning. These processes are frequently stereoselective.
- Plasma Protein Binding: The binding to proteins like human serum albumin (HSA) and α1-acid glycoprotein (AGP) can differ significantly between enantiomers. AGP, in particular, shows a pronounced affinity for basic drugs and often exhibits stereoselective binding. When one enantiomer is more extensively protein-bound, its free (unbound) concentration—the pharmacologically active fraction—is reduced, which can directly impact its distribution and efficacy [145].
- Tissue Distribution: Uptake into specific tissues may also be stereoselective if it involves binding to chiral tissue components or active transport processes.
Metabolism
Metabolism is the ADME process most frequently subject to stereoselectivity. The enzymes responsible for drug metabolism, primarily the cytochrome P450 (CYP) superfamily, are chiral proteins that can exhibit a strong preference for one enantiomer over the other.
The table below summarizes key examples of stereoselective metabolism:
Table 1: Case Studies in Stereoselective Metabolism and Pharmacokinetics
| Drug (Racemate) | Active Enantiomer | Metabolic & PK Differences | Clinical Implication |
|---|---|---|---|
| Warfarin [145] | S-warfarin (3-5x more potent) | S-warfarin metabolized mainly by CYP2C9; R-warfarin by CYP3A4/CYP1A2. Different clearance rates and half-lives. | Dosing complexity; drug interactions affecting CYP2C9 (e.g., sulfonamides) significantly alter anticoagulant effect. |
| Omeprazole [145] | Both S- and R- are active | R-omeprazole is rapidly metabolized by CYP2C19; S-omeprazole (Esomeprazole) is metabolized more slowly by CYP3A4. | Pure S-enantiomer (Esomeprazole) provides higher, more consistent plasma levels, allowing for lower and more predictable dosing. |
| Ibuprofen [145] | S-(+)-ibuprofen | R-(-)-ibuprofen is inactive at COX enzyme but undergoes enzymatic chiral inversion to the active S-form in the body. | Racemate is effective, but pure active S-form (Dexibuprofen) can provide faster onset at a lower dose. Inversion rate varies by ethnicity. |
| Propranolol [145] | S-(-)-propranolol | S-enantiomer is a potent β-blocker. The R-enantiomer is ~100-fold less active at β-receptors. | Early racemic formulation meant patients were exposed to a therapeutically largely inactive enantiomer. |
Several scenarios can occur with stereoselective metabolism:
- Pro-drug Activation: The distomer is the active compound, or a pro-drug that must be metabolically activated to the eutomer, as seen with ibuprofen.
- Divergent Metabolic Pathways: Each enantiomer is metabolized by a different enzyme system, leading to independent and unpredictable pharmacokinetics, as with warfarin.
- Chiral Inversion: The unidirectional bio-conversion of one enantiomer to its mirror image, a process exemplified by profens like ibuprofen.
- Genetic Polymorphisms: The impact of genetic variations in metabolizing enzymes (e.g., CYP2C19 poor metabolizers) can be enantiomer-specific, adding another layer of complexity to personalized dosing [145].
Excretion
The final elimination of drugs and their metabolites, often via renal or biliary excretion, can also be stereoselective. If renal excretion involves active secretion or reabsorption by chiral transporters in the proximal tubule, the clearance rates of enantiomers will differ. For instance, the renal clearance of S-(-)-atenolol has been reported to be higher than that of R-(+)-atenolol.
Experimental Protocols for Investigating Enantiomer PK
Studying the comparative pharmacokinetics of enantiomers requires specialized analytical and biological methods that can distinguish between the two mirror-image forms.
Analytical Techniques for Enantioseparation and Quantification
The cornerstone of enantiomer PK studies is the ability to separately quantify each enantiomer in complex biological matrices like plasma, urine, and tissues.
- Chiral Chromatography: This is the most widely used technique. It involves using high-performance liquid chromatography (HPLC) or ultra-performance liquid chromatography (UPLC) systems equipped with a chiral stationary phase (CSP). The CSP is designed with chiral selectors (e.g., cyclodextrins, proteins, macrocyclic glycopeptides) that transiently and selectively interact with each enantiomer, causing them to elute at different retention times. This allows for the individual quantification of each enantiomer when coupled with a detector (e.g., mass spectrometry, UV).
- Chiral Capillary Electrophoresis (CCE): This technique separates enantiomers based on their differential migration in an electric field when a chiral selector is added to the background electrolyte. It offers high separation efficiency and low solvent consumption.
In Vitro ADME Assays
Before proceeding to costly clinical trials, in vitro systems are used to screen for stereoselectivity.
- Metabolic Stability: Incubation of individual enantiomers with hepatocytes or liver microsomes from humans or preclinical species to determine their intrinsic clearance and identify which CYP enzymes are involved [146].
- Plasma Protein Binding: Techniques like ultrafiltration or equilibrium dialysis are used with racemic and individual enantiomers to measure the free fraction and identify stereoselective binding [146].
- Cell-Based Uptake/Efflux Assays: Using cell lines overexpressing specific transporters (e.g., P-gp, BCRP, OATPs) to assess if the enantiomers are substrates for chiral transport processes [146].
In Vivo Clinical Pharmacokinetic Studies
The definitive assessment of enantiomer PK is conducted in humans.
- Study Design: A clinical study is conducted where either the racemate or the individual enantiomers are administered to healthy volunteers or patients.
- Sample Collection: Serial blood samples are collected over time to generate a plasma concentration-time profile for each enantiomer separately.
- Data Analysis: Non-compartmental analysis (NCA) is used to calculate key PK parameters (AUC, C~max~, t~max~, t~1/2~, CL) for each enantiomer. For a more sophisticated analysis, Population PK (PopPK) modeling or Physiologically Based PK (PBPK) modeling can be employed to understand and predict the sources of variability in enantiomer exposure [147].
The following diagram illustrates a typical integrated workflow for evaluating the pharmacokinetics of enantiomers from early research to clinical application.
Diagram 1: Experimental Workflow for Enantiomer PK Assessment
Advanced Modeling and Machine Learning Approaches
Traditional experimental approaches are now being augmented by powerful in silico modeling techniques that can predict stereoselective ADME properties, accelerating the drug development process.
- Model-Informed Drug Development (MIDD): This is a framework that uses quantitative modeling and simulation to inform drug development and regulatory decision-making. For chiral drugs, MIDD tools can be applied from discovery through post-market surveillance [148].
- Quantitative Structure-Activity Relationship (QSAR): Computational models can predict the biological activity and ADME properties of enantiomers based on their chemical structures, helping prioritize candidates for synthesis and testing [148].
- Physiologically Based Pharmacokinetic (PBPK) Modeling: PBPK models are mechanistic tools that simulate the absorption, distribution, metabolism, and excretion of drugs in the human body. A PBPK model can be developed for individual enantiomers to predict their concentration-time profiles in plasma and tissues, and to assess the potential for drug-drug interactions [149] [148].
- Artificial Intelligence and Machine Learning (AI/ML): AI and ML are revolutionizing the field. As highlighted in recent research, "stereochemistry-aware generative models" can now design molecules with optimal 3D arrangements for desired properties [23]. Furthermore, AI can predict key ADME parameters directly from a compound's structural formula, which can then be fed into PBPK models—an approach known as AI-PBPK modeling [149]. This is particularly valuable in early discovery when experimental data is scarce. ML is also being used to automate traditionally labor-intensive processes like population PK model development, making the analysis of complex enantiomer data more efficient and reproducible [147].
Table 2: The Scientist's Toolkit: Key Reagents and Technologies for Enantiomer PK Research
| Tool/Reagent | Function in Enantiomer PK Research |
|---|---|
| Chiral Stationary Phases (CSPs) | Used in chiral HPLC/UPLC to physically separate enantiomers for individual quantification. |
| Cryopreserved Hepatocytes [146] | In vitro system to assess stereoselective metabolic stability and metabolite identification. |
| Human Liver Microsomes | Sub-cellular fraction containing CYP enzymes; used to determine enzyme-specific metabolic rates for each enantiomer. |
| Transfected Cell Lines | Engineered cells overexpressing specific human transporters (e.g., OATP1B1, P-gp) to study stereoselective uptake/efflux. |
| AI-PBPK Platforms [149] | Web-based simulators (e.g., B2O Simulator) that integrate machine learning with PBPK models to predict enantiomer PK/PD from structure. |
| Automated PopPK Software [147] | Tools (e.g., pyDarwin) that use optimization algorithms to automate the development of population PK models for clinical enantiomer data. |
The comparative pharmacokinetics of enantiomers is a sophisticated field grounded in the fundamental principles of stereochemistry. Significant differences in the ADME properties of enantiomers are the rule rather than the exception, driven by stereoselective interactions with the chiral biological environment. These differences can profoundly impact a drug's efficacy, safety, and dosing regimen.
The case studies of warfarin, omeprazole, and ibuprofen demonstrate that understanding these differences is not just an academic pursuit but a critical component of modern pharmacology. The historical practice of developing racemates is increasingly being replaced by the rational development of single-enantiomer drugs, a process supported by advanced analytical techniques, robust in vitro systems, and sophisticated in silico modeling approaches like PBPK and AI/ML. As machine learning models become increasingly "stereochemistry-aware," the drug discovery pipeline will become more efficient, enabling the precise design and development of optimal chiral therapeutics [23]. Therefore, a deep understanding of enantiomer pharmacokinetics remains indispensable for any researcher or organization committed to delivering safe and effective medicines.
Biological activity profiling is a cornerstone of modern drug discovery, providing critical insights into how small molecules interact with their biological targets. This profiling encompasses the comprehensive evaluation of target engagement—the binding of a compound to its intended protein target—and receptor specificity—the selectivity of this interaction across related proteins. Within the broader context of stereochemistry and isomerism research, understanding the three-dimensional spatial arrangement of atoms in a molecule becomes paramount, as stereoisomers can exhibit dramatically different biological activities despite identical molecular formulas and atomic connectivity [23]. The significance of stereochemistry is profoundly evident in drug discovery, where the spatial arrangement of atoms can significantly influence a compound's pharmacological properties, including binding affinity to target proteins, metabolic stability, and toxicity profiles [23]. For instance, the synthesis of methadone produces racemic mixtures of enantiomers, (R)-methadone and (S)-methadone. While (R)-methadone acts as an opioid for pain relief, (S)-methadone binds to the hERG protein and can lead to severe side effects, such as cardiac arrest [23]. This underscores the critical need for precise stereochemical characterization in biological activity profiling.
The challenges in this field are multifaceted. Conventional approaches for target identification, such as peptide microsequencing, immunohistochemical analysis, and degradation mass spectrometry (MS), while effective for identifying purified target proteins, have significant limitations [150]. Techniques like NMR experiments require high protein concentrations and may not accurately reflect the in-cell vulnerability of proteins, nor do they adequately quantify the potency of strongly binding drugs [150]. Furthermore, the increasing prominence of complex targets, including protein-protein interactions and allosteric binding sites, demands more sophisticated profiling technologies. This technical guide provides an in-depth examination of the core principles, methodologies, and data analysis techniques essential for rigorous biological activity profiling, with particular emphasis on the implications of molecular stereochemistry.
Methodologies for Assessing Target Engagement
Affinity-Based Protein Profiling (AfBPP)
Affinity-based protein profiling (AfBPP) has emerged as a fundamental method in chemical proteomics, leveraging molecular probes to assess the functional states of target proteins directly within biological systems [150]. This methodology significantly enhances our comprehension of disease mechanisms and facilitates the determination of potential therapeutic targets. Unlike conventional techniques, AfBPP enables researchers to detect target activity both in vivo and in situ, representing a significant breakthrough in drug discovery and biological research [150]. A prominent example of its application was the identification of the E3 ubiquitin ligase RNF114 as the direct target of the anti-cancer natural product Nimbolide, which was achieved using activity-based protein profiling (ABPP), a technique closely related to AfBPP [150].
AfBPs generally consist of three key components, each serving a distinct function in the profiling process [150]:
- Reactive Moieity: A functional group capable of binding to the target protein. This binding is typically non-covalent and reversible, which often has less impact on the protein's natural biological activities compared to covalent probes [150].
- Linker: A designed spacer that attenuates the effect of the label on the ligand's activity and provides appropriate spatial separation between the reactive moiety and the reporting tag [150].
- Label: A tag used for the detection or purification of the target protein. Common labels include biotin for affinity purification and fluorophores for visual detection [150].
Table 1: Major Categories of Affinity-Based Probes and Their Applications
| Probe Type | Key Characteristics | Primary Applications | Advantages | Limitations |
|---|---|---|---|---|
| Biotin Probes | Utilizes the high-affinity biotin-avidin system (BAS) for detection and amplification [150]. | Target identification and purification; immunology applications [150]. | High sensitivity and specificity; stable detection; single-step protocol [150]. | Susceptible to interference from free biotin in samples, potentially causing false-low results [150]. |
| FITC Probes | Employs fluorescein isothiocyanate (FITC) as a fluorescent label [150]. | Fluorescent detection and imaging of target proteins [150]. | Enables visualization and tracking without secondary reagents [150]. | Specific limitations not detailed in search results. |
| BRET Probes | Based on Bioluminescence Resonance Energy Transfer; uses NanoLuc luciferase as a donor [150]. | Monitoring target engagement and protein-protein interactions in live cells [150]. | Allows for real-time, dynamic monitoring in a live-cell environment [150]. | Requires genetic engineering for tag incorporation [150]. |
| Radiolabeled Probes | Incorporates radioactive isotopes for detection [150]. | Highly sensitive quantification and binding studies [150]. | Offers extremely high sensitivity for detection and quantification [150]. | Handling and disposal of radioactive materials require special safety protocols [150]. |
Experimental Protocol: AfBPP for Live Cells
The following protocol details a standard workflow for using AfBPs to identify cellular protein targets:
- Cell Culture and Preparation: Culture adherent or suspension cells in appropriate medium under standard conditions (e.g., 37°C, 5% CO₂). On the day of the experiment, harvest cells and seed them at a desired density in multi-well plates or culture flasks.
- Probe Incubation: Prepare a stock solution of the AfBP in a suitable solvent (e.g., DMSO). Dilute the probe to the desired working concentration in serum-free or complete culture medium. Remove the growth medium from cells, add the probe-containing medium, and incubate for the predetermined time (typically 1-4 hours) under standard culture conditions.
- Cell Lysis: After incubation, remove the probe-containing medium and wash the cells gently with phosphate-buffered saline (PBS). Lyse the cells using an appropriate lysis buffer (e.g., RIPA buffer supplemented with protease and phosphatase inhibitors) on ice for 15-30 minutes. Scrape the lysates and transfer them to microcentrifuge tubes.
- Centrifugation: Clarify the cell lysates by centrifugation at high speed (e.g., 14,000 × g for 15 minutes at 4°C). Collect the supernatant, which contains the solubilized proteins.
- Affinity Purification (For biotin-labeled probes): Incubate the clarified lysates with streptavidin- or avidin-conjugated beads for 1-2 hours at 4°C with gentle rotation. This step captures the biotin-tagged probe and its bound target proteins.
- Washing: Pellet the beads by gentle centrifugation and wash them extensively with lysis buffer to remove non-specifically bound proteins.
- Elution and Analysis:
- For Mass Spectrometry (MS) Analysis: The captured protein complexes can be digested on-beads with trypsin. The resulting peptides are then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for protein identification.
- For Western Blot Analysis: Bound proteins can be eluted from the beads by boiling in SDS-PAGE loading buffer. The eluates are then separated by gel electrophoresis and transferred to a membrane for immunoblotting with specific antibodies.
Diagram 1: AfBPP Workflow for Live Cells
Quantitative Analysis of Receptor Specificity
Quantitative assessment of receptor specificity is crucial for understanding a compound's mechanism of action and predicting potential off-target effects. This involves measuring binding affinities and functional responses across a panel of related receptors. Quantitative data analysis methods, which focus on measurable information such as counts, percentages, and averages, are essential for summarizing datasets, identifying relationships between variables, and making predictions in this context [151]. Key statistical techniques include descriptive statistics (e.g., mean, median, standard deviation) to summarize data and inferential statistics (e.g., t-tests, ANOVA, regression analysis) to make generalizations and test hypotheses about a larger population based on sample data [151].
A powerful application of structure-function studies is the engineering of receptor specificity and activity. Research on the chemokines CCL27 and CCL28, which share the receptor CCR10, demonstrates how N-terminal residues are critical determinants of receptor pharmacology [152]. For instance, a mere two-amino-acid deletion from the N-terminus of CCL27 resulted in a CCR10 antagonist, while extension with a single phenylalanine produced a CCR10 superagonist by occupying a unique subpocket in the receptor [152]. Furthermore, swapping the N-terminus of CCL28 onto the globular domain of CCL27 created a superagonist for CCR10, whereas the reciprocal swap showed reduced activity, indicating that the CCL28 N terminus is a stronger driver of CCR10 signaling [152]. AlphaFold models of these complexes help rationalize these functional outcomes and reveal the structural basis for specificity, such as the reduced size and more basic nature of the binding pocket of CCR3, which contributes to its exclusive specificity for CCL28 over CCL27 [152].
Table 2: Quantitative Functional Data for CCL27 and CCL28 Mutants
| Chemokine Variant | Receptor | Signaling Efficacy (Relative to WT) | Key Functional Outcome | Structural Rationale |
|---|---|---|---|---|
| [3-88]-CCL27 (Truncation) | CCR10 | Extremely low-efficacy partial agonist [152] | CCR10 antagonist; blocks cell migration to WT CCL27 [152] | Loss of critical interactions in transmembrane pocket [152] |
| F1A-CCL27 | CCR10 | ~35% reduction in efficacy, 10-fold reduction in potency [152] | Reduced cell migration [152] | Phe1 directly interacts with Tyr in TM binding pocket [152] |
| L2A-CCL27 | CCR10 | ~15-20% reduction in migration at 50nM [152] | Moderately reduced cell migration [152] | Leu2 is important for activation but less critical than Phe1 [152] |
| NT28-CCL27 (Chimera) | CCR10 | Increased activity (Superagonist) [152] | Enhanced CCR10 signaling [152] | CCL28 N terminus is a stronger driver of CCR10 signaling [152] |
| CCL28 | CCR10 & CCR3 | Equivalent or reduced for NT27-CCL28 swap [152] | Activates both CCR10 and CCR3 [152] | Basic N terminus and ECLs of CCR3 accommodate CCL28 [152] |
The following diagram illustrates the key logical relationships and structural determinants that govern receptor specificity and engagement, as revealed by studies on chemokines like CCL27 and CCL28.
Diagram 2: Determinants of Receptor Specificity
The Scientist's Toolkit: Essential Reagents and Materials
Successful biological activity profiling relies on a suite of specialized reagents and tools. The following table details key solutions and their applications in experiments for target engagement and receptor specificity.
Table 3: Research Reagent Solutions for Activity Profiling
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Affinity-Based Probes (AfBPs) | Label and isolate target proteins in complex biological mixtures via a reactive moiety, linker, and tag [150]. | Identify cellular targets of a novel small molecule drug candidate. |
| Biotin-Streptavidin System | High-affinity system used for purification and detection of biotin-tagged probes and their bound targets [150]. | Pull-down and concentrate probe-bound protein complexes from cell lysates for MS analysis. |
| AlphaFold Models | AI-powered computational models predicting the 3D structure of protein-ligand complexes [152]. | Rationalize the functional impact of mutations (e.g., N-terminal swaps) on receptor signaling. |
| Stable Cell Lines | Cells engineered to stably express a receptor or target protein of interest. | Generate reproducible datasets for receptor activation and internalization assays. |
| Proteomics-Grade Lysis Buffer | Buffer designed to solubilize proteins while maintaining interactions and inhibiting proteases. | Prepare clear, protein-rich lysates from probe-treated cells for affinity purification. |
| LC-MS/MS System | Analytical platform for separating, identifying, and quantifying proteins from complex mixtures. | Identify unknown protein targets captured by an AfBP. |
| Activity-Based Probes (AcBPs) | Covalently label the active sites of enzymes (e.g., serine hydrolases, proteases) to report on their functional state [150]. | Profile enzyme activity changes in disease models or in response to drug treatment. |
Biological activity profiling, integrating sophisticated methodologies like affinity-based protein profiling and quantitative functional assays, provides an indispensable framework for understanding the molecular interactions underpinning drug action. The integration of stereochemical considerations and AI-powered structural modeling, as exemplified by AlphaFold, is refining our ability to predict and engineer receptor specificity and pharmacological efficacy [152] [23]. As the field advances, the development of more sensitive, reversible probes and the creation of increasingly realistic, stereochemistry-aware computational benchmarks will be crucial for identifying novel therapeutic targets and accelerating the development of safer, more effective drugs with precise target engagement profiles [150] [23]. The future of activity profiling lies in its increasing integration with structural biology and computational chemistry, enabling a truly rational design of molecules with desired biological activities.
Metabolic Pathway Variations Between Stereoisomers
Stereoisomers are molecules with identical atomic constitution and bonding but differ in the three-dimensional arrangement of their atoms [153]. Among these, enantiomers—mirror-image stereoisomers around one or more asymmetric chiral centers—are of profound importance in biological systems [3]. Despite nearly identical physical and chemical properties in achiral environments, enantiomers exhibit distinct behaviors in chiral biological milieus, leading to significant differences in their pharmacokinetics, pharmacodynamics, and metabolic fates [3] [153]. This divergence arises because enzymes, receptors, and transporters in living organisms are themselves chiral and can interact differently with each member of an enantiomeric pair [3]. Consequently, the metabolic pathways of stereoisomers often vary considerably, influencing their biological activity, toxicity, and environmental persistence.
Understanding these metabolic variations is critical across multiple fields. In pharmaceutical sciences, it underpins drug safety and efficacy, as enantiomers may be metabolized at different rates or via different pathways, leading to distinct active or toxic metabolites [3] [153]. In environmental science, the stereospecific biodegradation of pollutants by microorganisms determines their persistence and ecological impact [154]. In plant biology, the evolution of stereospecific enzymes drives the diversity of specialized metabolites with distinct ecological roles [155]. This guide synthesizes current research to provide an in-depth analysis of the mechanisms, analytical methods, and biological implications of metabolic pathway variations between stereoisomers, framed within the broader context of stereochemistry research.
Fundamental Mechanisms of Stereospecific Metabolism
Molecular Basis for Stereospecific Recognition
The primary mechanism driving metabolic variations between stereoisomers is the differential binding to chiral biological macromolecules. As illustrated in Figure 1, a chiral binding site preferentially accommodates one enantiomer over the other due to spatial constraints and interaction requirements [3]. For a drug or metabolite to exert its effect, specific portions of the molecule (labeled A, B, C) must align precisely with corresponding regions (a, b, c) of the binding site. The active enantiomer fits this binding site perfectly, allowing all necessary interactions. In contrast, the inactive enantiomer cannot achieve this optimal alignment, no matter how it is rotated in space, preventing effective binding and subsequent biological response [3]. This molecular recognition principle extends to enzyme-active sites, transporters, and receptors, ultimately governing the stereospecificity of metabolic transformations.
Key Enzymes and Metabolic Fates
Stereospecific metabolism is often mediated by enzymes that exhibit strong enantioselectivity. Cytochrome P450 enzymes (CYPs) frequently display distinct catalytic efficiency toward different enantiomers, leading to variations in metabolic rates and pathways [154] [155]. For instance, the cytochrome P450 enzyme GAME8 (CYP72A208) in Solanum species hydroxylates the terminal methyl group of cholesterol with absolute stereospecificity, producing either the 25S or 25R isomer in different plants [155]. Dehalogenases, such as LinB, catalyze stereospecific biotransformation of pollutants like hexabromocyclododecane (HBCD) [154]. Lactate dehydrogenases (LDHs) distinguish between L- and D-lactate isomers, linking stereospecific metabolism to post-translational modifications like lactylation [156].
The metabolic fates of stereoisomers diverge through several pathways:
- Biotransformation Rate Differences: One enantiomer may be metabolized rapidly while the other persists.
- Alternative Pathway Activation: Different enantiomers may undergo distinct metabolic routes yielding unique metabolite profiles.
- Product Stereochemistry Variations: Enzymatic reactions may produce stereochemically distinct metabolites from different substrate enantiomers.
- Post-Translational Modification Specificity: Metabolites like L- and D-lactate drive stereospecific modifications (KL-la and KD-la) on proteins, influencing gene expression and cellular function [156].
Case Studies of Stereospecific Metabolic Pathways
Microbial Biotransformation of Environmental Pollutants
The flame retardant hexabromocyclododecane (HBCD) exists as multiple stereoisomers, and its environmental fate demonstrates pronounced stereospecific metabolism. Acinetobacter hemolyticus sp. strain HW-2 transforms HBCD stereoisomers with remarkable efficiency and selectivity [154]. As shown in Table 1, the bacterium exhibits varying transformation efficiencies for different HBCD enantiomers within just three days of exposure.
Table 1: Stereospecific Biotransformation of HBCD Isomers by Strain HW-2
| HBCD Stereoisomer | Transformation Efficiency (%) |
|---|---|
| (+)-α-HBCD | 52.38 |
| (-)-α-HBCD | 71.08 |
| (+)-β-HBCD | 71.07 |
| (-)-β-HBCD | 63.34 |
| (+)-γ-HBCD | 47.47 |
| (-)-γ-HBCD | 77.05 |
This stereospecificity arises from differential expression of catabolic genes. Transcriptomic analysis revealed distinct gene expression patterns in strain HW-2 when exposed to different HBCD enantiomers [154]. Key enzymes involved in this stereospecific biotransformation include hydrolytic dehalogenases, glutathione-S-transferases (GST), and cytochrome P450 enzymes, which are upregulated to varying degrees depending on the stereoisomer present [154]. The environmental implication is significant: the preferential transformation of γ-HBCD over α-HBCD explains why α-HBCD becomes the predominant diastereomer in biotic media despite being a minor component in technical HBCD mixtures [154].
Lactate Stereoisomers in Bacterial and Human Metabolism
Lactate exists as two stereoisomers, L-lactate and D-lactate, that undergo distinct metabolic pathways with important biological consequences. L-lactate is predominantly produced by mammalian cells via L-lactate dehydrogenase (L-LDH) during anaerobic glycolysis, while D-lactate is primarily generated by bacterial metabolism through D-lactate dehydrogenase (D-LDH) or the methylglyoxal pathway [156]. These stereoisomers drive different post-translational modifications: L-lactate mediates L-lactylation (KL-la) via enzymatic reactions involving specific "writer" (e.g., p300/CBP, GCN5) and "eraser" (e.g., HDAC1-3, SIRT1-3) enzymes [156]. In contrast, D-lactate forms D-lactylation (KD-la) non-enzymatically from lactoylglutathione (LGSH), a product of the glyoxalase pathway [156].
Table 2: Metabolic Characteristics of Lactate Stereoisomers
| Parameter | L-Lactate | D-Lactate |
|---|---|---|
| Primary Source | Mammalian cells, anaerobic glycolysis | Bacterial metabolism |
| Forming Enzyme | L-lactate dehydrogenase (L-LDH) | D-lactate dehydrogenase (D-LDH) |
| Metabolic Pathway | Glycolysis | Glycolysis, methylglyoxal pathway |
| Lactylation Type | KL-la (enzymatic) | KD-la (non-enzymatic) |
| Associated Diseases | Metabolic acidosis, neurodegenerative diseases | D-lactic acidosis, short-bowel syndrome |
These metabolic differences have direct clinical relevance. D-lactate accumulation in conditions like short-bowel syndrome can cause D-lactic acidosis, while L-lactate buildup is associated with metabolic acidosis and neurodegenerative diseases [156]. The stereospecific lactylation modifications influence gene expression, immune responses, and pathogen virulence, demonstrating how metabolic variations between stereoisomers can shape host-pathogen interactions and disease progression [156].
Stereochemical Diversity in Plant Alkaloid Biosynthesis
The Solanum genus (including tomato, potato, and eggplant) exhibits fascinating stereochemical diversity in steroidal glycoalkaloid (SGA) biosynthesis, particularly at the C25 position [155]. Different species produce either 25S or 25R stereoisomers of these defense compounds, controlled by stereospecific cytochrome P450 enzymes from the GAME8 family [155]. Phylogenetic analysis reveals that GAME8 enzymes cluster into two distinct clades: a "tomato clade" that produces 25S isomers and an "eggplant clade" that produces 25R isomers [155]. This stereospecificity arises from the enzyme's regioselectivity in hydroxylating one of the two terminal methyl groups (C26 or C27) of cholesterol, creating either the 25S or 25R chiral center [155].
The evolutionary history of these enzymes demonstrates how metabolic pathways diversify to generate stereochemical variation. Ancestral GAME8 likely favored the 25R configuration, with gene duplications giving rise to 25S-producing enzymes in more recent Solanum species [155]. In a remarkable case of reverse evolution, wild S. cheesmaniae from the Galápagos Islands has mutations in GAME8 that caused a reversion from 25S back to the ancestral 25R configuration [155]. This stereochemical variation affects the biological activity of SGAs and represents an evolutionary adaptation in plant defense mechanisms [155].
Analytical Methods for Studying Stereospecific Metabolism
Experimental Protocols for Stereospecific Biotransformation Studies
Investigating metabolic pathway variations between stereoisomers requires specialized experimental approaches that account for chiral complexity.
Protocol 1: Assessing Stereospecific Microbial Transformation
- Stereoisomer Preparation: Separate chiral isomers using preparative HPLC with chiral stationary phases. Manually collect individual enantiomers at concentrations >7 mg/L for subsequent experiments [154].
- Biotransformation Assay: Inoculate the microbial strain (e.g., Acinetobacter hemolyticus sp. strain HW-2) into minimal salt medium containing a specific stereoisomer as the sole carbon source [154].
- Sampling and Extraction: Collect culture samples at predetermined intervals. Extract residual substrate and metabolites using organic solvents like methanol or acetonitrile [154].
- Chiral Analysis: Quantify stereoisomer depletion and metabolite formation using chiral liquid chromatography-mass spectrometry (LC-MS). Monitor transformation efficiency over time [154].
- Transcriptomic Analysis: Extract total RNA from cells exposed to different stereoisomers. Perform RNA sequencing and differential gene expression analysis to identify stereospecific metabolic pathways [154].
Protocol 2: Characterizing Plant Enzyme Stereospecificity
- Gene Cloning: Isolate GAME8 orthologs from different Solanum species (e.g., tomato SlGAME8 and eggplant SmGAME8) via PCR amplification [155].
- Heterologous Expression: Co-infiltrate Nicotiana benthamiana leaves with GAME8 genes along with other pathway genes (GAME15, GAME6, GAME11, GAME12, GAME4) using Agrobacterium tumefaciens-mediated transformation [155].
- Metabolite Analysis: Harvest leaf tissue after 5-7 days. Extract metabolites and analyze aglycone profiles using UPLC-TOF-MS [155].
- Stereochemical Assignment: Identify C25 configuration of products by comparing retention times and fragmentation patterns with authentic standards (e.g., tomatidenol for 25S, solasodine for 25R) [155].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Stereospecific Metabolism Studies
| Reagent/Technique | Function in Stereospecific Studies |
|---|---|
| Chiral HPLC Columns | Separation and purification of individual stereoisomers for metabolic experiments [154] |
| Stable Isotope-Labeled Standards | (e.g., 13C12-labeled HBCD isomers) Quantification of stereoisomer depletion and metabolite formation [154] |
| Chiral LC-MS/MS | Sensitive detection and quantification of stereoisomers and their metabolites in complex matrices [154] |
| qRT-PCR Assays | Measurement of stereospecific gene expression responses to different enantiomers [154] |
| Heterologous Expression Systems | (e.g., N. benthamiana, E. coli) Functional characterization of stereospecific enzymes [155] |
| Chiral Synthetic Intermediates | (e.g., L/D-lactate, chiral aglycones) Substrates for in vitro enzyme assays and metabolic tracing [156] |
Statistical Considerations for Metabolomics Data
Analyzing stereospecific metabolism often generates complex, high-dimensional data requiring specialized statistical approaches. With an increasing number of metabolites assayed, multivariate methods like sparse partial least squares (SPLS) and least absolute shrinkage and selection operator (LASSO) outperform traditional univariate methods [157]. These approaches better handle the high intercorrelation between metabolites, reducing false discovery rates in nontargeted metabolomics datasets where the number of metabolites may exceed the number of study subjects [157]. For smaller sample sizes with targeted metabolite panels, univariate methods with false discovery rate (FDR) correction remain effective [157].
Regulatory and Drug Development Perspectives
Regulatory agencies worldwide recognize the importance of stereospecific metabolism in drug development. The U.S. Food and Drug Administration's 1992 policy states that "the stereoisomeric composition of a drug with a chiral center should be known and the quantitative isomeric composition of the material used in pharmacologic, toxicologic, and clinical studies known" [153]. Key requirements include:
- Chiral Assay Development: Quantitative assays for individual enantiomers in biological samples must be available early in drug development to assess potential interconversion and stereospecific pharmacokinetics [153].
- Stereochemical Characterization: Applications for enantiomeric drugs must include stereochemically specific identity tests and stereochemically selective assay methods [153].
- Specifications and Controls: Manufacturers must assure identity, strength, quality, and purity from a stereochemical viewpoint, with appropriate limits for isomeric impurities [153].
When stereoisomers are biologically distinguishable, they should be considered different drugs, as they may have different pharmacokinetic properties and quantitatively or qualitatively different pharmacologic or toxicologic effects [153]. For racemic drugs, manufacturers must monitor enantiomers individually when their pharmacokinetic profiles differ to assess dose linearity, drug interactions, and effects of metabolic or excretory impairment [153].
Visualizing Stereospecific Metabolic Pathways
Diagram 1: Stereospecific Enzyme Recognition. The chiral enzyme preferentially binds and metabolizes the S-enantiomer over the R-enantiomer due to optimal spatial compatibility.
Diagram 2: Microbial Stereospecific Biodegradation Workflow. Bacterial exposure to HBCD stereoisomers triggers differential gene expression of catabolic enzymes, leading to stereospecific degradation patterns.
Metabolic pathway variations between stereoisomers represent a fundamental principle with far-reaching implications across pharmaceutical sciences, environmental biotechnology, and chemical ecology. The case studies presented—ranging from microbial degradation of environmental pollutants to stereospecific alkaloid biosynthesis in plants and lactate metabolism in humans—demonstrate that stereochemistry is not a subtle nuance but rather a determinant factor in metabolic fate and biological activity. Understanding these variations requires sophisticated analytical methods, including chiral separations, stereospecific metabolomics, and multivariate statistical analyses. As regulatory guidelines continue to emphasize thorough stereochemical characterization, particularly in drug development, research into stereospecific metabolism will remain crucial for designing safer pharmaceuticals, predicting environmental fate of pollutants, and understanding evolutionary adaptations in natural product biosynthesis. The integration of stereochemical considerations into metabolic studies represents an essential approach for advancing both basic science and applied technologies across multiple disciplines.
The therapeutic index (TI) is a fundamental quantitative measurement in pharmacology that defines the relative safety of a drug by comparing the amount that causes a toxic effect to the amount that elicits the desired therapeutic response [158]. This critical parameter, also referred to as the therapeutic ratio, provides researchers and clinicians with a numerical value representing the margin of safety between efficacy and toxicity. For any drug development program, accurately determining the TI is essential for establishing dosing regimens that maximize therapeutic benefit while minimizing adverse effects.
Classically, TI is expressed as the ratio of the dose that produces a toxic effect in 50% of the population (TD50) to the dose that produces a therapeutic effect in 50% of the population (ED50): TI = TD50/ED50 [158] [159]. In preclinical animal studies, the median lethal dose (LD50) is often substituted for TD50, giving TI = LD50/ED50 [160] [161]. The related concept of the therapeutic window refers to the range of doses between the minimum effective concentration and the maximum tolerated concentration, providing a practical dosing range for clinical use [158].
For drugs intended for human use, the determination of TI has evolved beyond simple animal lethality studies. Modern drug development utilizes plasma exposure levels (e.g., area under the concentration-time curve) to calculate TI, providing a more clinically relevant safety assessment that accounts for inter-individual variability in drug metabolism and disposition [158]. This approach is particularly important for drugs with complex pharmacokinetic profiles or those subject to significant metabolic polymorphisms.
Quantitative Foundations of Therapeutic Index
Core Definitions and Calculations
The quantitative assessment of therapeutic index relies on several key parameters derived from dose-response relationships:
- ED50 (Median Effective Dose): The dose that produces a specified therapeutic effect in 50% of the population [161]. This parameter represents the potency of a drug for its intended effect.
- TD50 (Median Toxic Dose): The dose required to produce a particular toxic effect in 50% of subjects [161]. This endpoint should reflect clinically relevant toxicity rather than arbitrary laboratory values.
- LD50 (Median Lethal Dose): The dose required to kill 50% of a test population, typically determined in animal studies [158] [161].
- Therapeutic Window: The dosage range between the minimum dose that provides therapeutic efficacy and the maximum dose that remains tolerable without significant toxicity [161].
Two primary types of therapeutic indices are recognized in modern pharmacology. The safety-based therapeutic index (TI = LD50/ED50) focuses on lethal effects, while the efficacy-based therapeutic index (TI = ED50/TD50) emphasizes the relationship between therapeutic and toxic (non-lethal) effects [158]. The protective index (TD50/ED50) is often more informative than traditional TI calculations since many drugs produce significant toxicity at doses far below those causing death [158].
Therapeutic Index Values for Representative Drugs
Table 1: Therapeutic Indices of Selected Pharmaceuticals
| Drug | Therapeutic Index | Clinical Implications |
|---|---|---|
| Remifentanil (Opioid analgesic) | 33,000:1 [158] | Very wide safety margin; forgiving dosing range |
| Diazepam (Benzodiazepine) | 100:1 [158] | Moderate safety margin |
| Morphine (Opioid analgesic) | 70:1 [158] | Requires careful dose titration |
| Cocaine (Stimulant, anesthetic) | 15:1 [158] | Narrow safety margin |
| Ethanol (Sedative) | 10:1 [158] | Narrow safety margin |
| Digoxin (Cardiac glycoside) | 2:1 [158] | Very narrow safety margin; requires monitoring |
Table 2: Narrow Therapeutic Index (NTI) Drugs and Monitoring Requirements
| NTI Drug | Therapeutic Area | Monitoring Parameters |
|---|---|---|
| Warfarin [158] [162] | Anticoagulation | INR (International Normalized Ratio) |
| Lithium [158] [162] | Psychiatry | Serum lithium levels |
| Theophylline [158] | Respiratory | Serum theophylline levels |
| Gentamicin [158] | Anti-infective | Trough and peak serum levels |
| Vancomycin [158] | Anti-infective | Trough serum levels |
Drugs with a narrow therapeutic index (NTI), typically defined as those with less than a twofold difference between median toxic and effective concentrations, require particularly careful management [162]. For these agents, small changes in dose or blood concentration may lead to serious therapeutic failure or severe adverse drug reactions [162]. Regulatory agencies have established special requirements for NTI drugs, including tightened bioequivalence limits for generic versions, often requiring a full-replicate, crossover bioequivalence study [162].
Experimental Protocols for TI Determination
Preclinical TI Assessment Workflow
Diagram 1: Preclinical TI Assessment
The experimental determination of therapeutic index begins with appropriate study design and model selection. For in vivo assessments, the choice of animal model should consider species-specific differences in drug metabolism and pathophysiology that might affect the translatability of results to humans [163]. Dose selection should encompass a sufficient range to characterize both the efficacy and toxicity dose-response curves adequately.
Efficacy testing involves administering the test compound across a range of doses to groups of animals and measuring the predetermined therapeutic endpoint. The ED50 is then calculated from the resulting dose-response curve using appropriate statistical methods (e.g., probit analysis, nonlinear regression) [160]. Similarly, toxicity testing involves administering the compound across a dose range and monitoring for predefined toxicological endpoints, with TD50 calculated from the resulting curve [160]. For acute toxicity assessments, LD50 may be determined, though modern approaches favor using more clinically relevant toxicological endpoints over lethality [158].
Advanced TI Determination Methodologies
Contemporary approaches to TI determination have evolved beyond traditional methods:
Exposure-Based TI Assessment: Modern drug development calculates TI using plasma exposure levels (e.g., AUC, Cmax) rather than administered dose, as it is the tissue exposure to drug over time that drives both pharmacological and toxicological effects [158]. This approach accounts for inter-individual variability in pharmacokinetics due to polymorphisms in metabolism, drug-drug interactions, or differences in body weight [158].
Genotype-Phenotype Difference (GPD) Framework: Advanced machine learning frameworks incorporate genotype-phenotype differences between preclinical models and humans to improve prediction of human drug toxicity [163]. This approach assesses differences in three biological contexts: gene essentiality, tissue expression profiles, and network connectivity of drug targets [163]. By incorporating these inter-species differences, this model demonstrated enhanced predictive accuracy (AUROC = 0.75 vs. baseline 0.50), particularly for neurotoxicity and cardiovascular toxicity [163].
Replicate Crossover Design for NTI Drugs: For drugs with narrow therapeutic indices, regulatory agencies recommend full-replicate, crossover bioequivalence studies where the same subject receives both the reference product and the test product twice [162]. This design allows simultaneous comparisons of mean pharmacokinetics and within-subject variability between products, which is critical for ensuring the safety of NTI drugs [162].
Stereochemistry and Isomerism in Therapeutic Index
Molecular Chirality and Biological Activity
Stereochemistry represents a critical dimension in therapeutic index optimization, as the spatial arrangement of atoms in a molecule can profoundly influence both its pharmacological activity and toxicological profile. Stereoisomers - compounds with the same molecular formula and atomic connectivity but different spatial arrangements - can exhibit markedly different biological properties despite their chemical similarity [23].
The importance of stereochemistry is particularly evident in drug discovery, where the spatial arrangement of atoms can significantly influence a compound's pharmacological properties, including binding affinity to target proteins, metabolic stability, and toxicity [23]. For example, the synthesis of methadone produces racemic mixtures of enantiomers - (R)-methadone and (S)-methadone. While (R)-methadone acts as an opioid for pain relief, (S)-methadone has been identified to bind to the hERG protein and can lead to severe side effects, such as heart attacks or cardiac arrest [23]. This dramatic difference in safety profile between enantiomers highlights why stereochemical considerations are essential for accurate therapeutic index determination.
Diagram 2: Stereochemistry in TI Assessment
Stereochemistry-Aware Drug Design
Recent advances in computational chemistry have enabled the development of stereochemistry-aware molecular generative models that explicitly account for three-dimensional molecular structure during the drug design process [23]. These models utilize string-based molecular representations (SMILES, SELFIES, or GroupSELFIES) that natively encode stereochemical information through specific tokens denoting chirality and E/Z geometric isomerism [23].
Studies comparing stereochemistry-aware models with conventional approaches have demonstrated that explicitly considering stereochemistry during molecular optimization improves performance on stereochemistry-sensitive tasks, including optimization of drug activity and safety profiles [23]. However, this approach increases the complexity of the chemical space that must be navigated, presenting computational challenges [23].
The impact of stereochemistry extends beyond pharmaceutical applications to materials science, where stereoisomerism in energetic materials based on 2,4,10-trioxaadamantane backbones resulted in measurable differences in density, stability, and performance despite identical molecular formulas and functional group positions [86]. This principle similarly applies to pharmaceutical compounds, where stereochemistry can influence crystal packing, solubility, and ultimately bioavailability and safety.
Research Tools and Methodologies
Essential Research Reagents and Solutions
Table 3: Key Research Reagents for TI Assessment
| Reagent/Category | Function/Application | Specific Examples |
|---|---|---|
| In Vitro CYP Enzyme Assays [164] | Metabolic stability assessment; drug interaction potential | CYP450 isoforms (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4/5) |
| Transporter Assay Systems [164] | Membrane permeability; drug distribution | P-gp, BCRP, OATP1B1, OATP1B3, OAT1 substrates |
| Cytotoxicity Assays [165] | Preliminary toxicity screening | CC50 (half-maximal cytotoxic concentration) determination |
| Selective CYP Inhibitors [164] | Reaction phenotyping; metabolic pathway identification | α-naphthoflavone (CYP1A2), sulfaphenazole (CYP2C9), quinidine (CYP2D6) |
| CYP Inducers [164] | Drug interaction studies; enzyme induction potential | Omeprazole (CYP1A2), rifampicin (multiple CYP enzymes) |
| Stable Isotope Labels | Metabolic profiling; quantitative analysis | Deuterated, C13, N15 labeled internal standards |
Analytical and Computational Tools
Modern therapeutic index assessment employs sophisticated analytical and computational platforms:
High-Resolution Mass Spectrometry: Essential for quantitative bioanalysis of drugs and metabolites in biological matrices. LC-MS/MS systems provide the sensitivity and specificity required for pharmacokinetic studies supporting exposure-based TI calculations.
Cellular Toxicity Screening Platforms: High-content screening systems combine automated microscopy with image analysis to assess multiple toxicity endpoints simultaneously, including cell viability, mitochondrial membrane potential, and oxidative stress.
In Silico Prediction Tools: Machine learning frameworks that incorporate chemical properties alongside biological data, such as the genotype-phenotype differences (GPD) model, which integrates inter-species differences in gene essentiality, tissue specificity, and network connectivity to predict human-specific drug toxicity [163].
Stereochemistry-Aware Generative Models: Computational tools like modified REINVENT and JANUS algorithms that incorporate stereochemical information during molecular generation and optimization, enabling more efficient exploration of stereochemically-defined chemical space [23].
The accurate determination of therapeutic index remains a cornerstone of pharmaceutical development, bridging preclinical discovery and clinical application. While traditional TI calculations based on median effective and toxic doses provide valuable safety estimates, modern approaches utilizing plasma exposure levels and incorporating inter-species differences in genotype-phenotype relationships offer enhanced predictive accuracy for human outcomes [158] [163].
The critical influence of stereochemistry on therapeutic index underscores the importance of considering three-dimensional molecular structure throughout drug optimization. As stereochemistry-aware generative models continue to evolve, they hold promise for more efficient design of therapeutic agents with optimized efficacy and safety profiles [23]. Furthermore, the integration of advanced machine learning approaches with high-throughput experimental data will likely enhance our ability to predict human-relevant therapeutic indices earlier in the development process, potentially reducing attrition rates and improving patient safety [163].
For researchers working with narrow therapeutic index drugs, stringent bioequivalence standards and therapeutic drug monitoring remain essential for ensuring patient safety, particularly as generic versions of complex drugs enter the market [162]. As our understanding of the interplay between efficacy, toxicity, and molecular structure deepens, therapeutic index assessments will continue to evolve, incorporating increasingly sophisticated approaches to balance therapeutic benefit against potential harm.
The efficacy of beta-lactam antibiotics, one of the most crucial classes of antimicrobial agents in clinical history, is profoundly influenced by their three-dimensional structure. Within the broader research on stereochemistry and isomerism in organic molecules, the absolute configuration of beta-lactam rings and their substituents dictates critical biological interactions, including binding affinity to penicillin-binding proteins (PBPs) and susceptibility to enzymatic degradation by beta-lactamases [166] [167]. The rise of antimicrobial resistance, driven by bacterial production of beta-lactamases that hydrolyze the essential beta-lactam ring, represents a severe global health threat [168] [169]. This case study explores how stereochemical principles are being leveraged in the design of next-generation beta-lactams and conjugates to overcome these resistance mechanisms, providing an in-depth technical analysis for researchers and drug development professionals.
Stereochemical Fundamentals of Beta-Lactam Activity
The biological activity of beta-lactam antibiotics is inherently tied to their chirality. The beta-lactam ring itself contains a stereogenic center, and additional chiral centers on the fused ring and side chains further define its three-dimensional structure.
- Absolute Configuration and PBP Binding: The interaction between beta-lactams and their target PBPs is highly stereospecific. The beta-lactam ring mimics the D-Ala-D-Ala terminus of peptidoglycan precursors. The stereochemistry of the lactam ring and its fused system determines how effectively it fits into the enzyme's active site, facilitating acylation and irreversible inhibition of the transpeptidase activity essential for bacterial cell wall synthesis [167]. Molecules with the incorrect configuration show drastically reduced binding affinity.
- Siderophore Conjugates and Chirality: Novel siderophore-beta-lactam conjugates (sideromycins) such as cefiderocol exploit iron-scavenging pathways for cellular entry. The synthetic siderophore moieties in these compounds, such as the bis-catechol in BAMP (azotochelin-linked ampicillin) and BLOR (azotochelin-linked loracarbef), or the mixed bis-catechol-mono-hydroxamate in MCEF (inspired by fimsbactin), are complex molecules that likely contain multiple chiral centers [170]. The stereochemistry of these siderophore components is critical for recognition by specific bacterial TonB-dependent transporters (TBDTs), governing uptake efficiency and spectrum of activity [170].
Stereochemistry governs multiple stages of beta-lactam antibiotic action, from membrane transport to target binding. TBDTs: TonB-dependent transporters.
Emerging Beta-Lactam Designs and Stereochemical Considerations
Research into overcoming resistance has yielded innovative beta-lactam designs where stereochemistry plays a pivotal role. These include siderophore conjugates, bis-beta-lactams, and combinations with novel beta-lactamase inhibitors.
Siderophore-Beta-Lactam Conjugates
Sideromycins represent a "Trojan horse" strategy, hijacking bacterial iron acquisition systems. Their enhanced activity is species-dependent and involves a complex interplay of factors [170].
Key Findings from Recent Research:
- Potency Enhancement: Conjugation of a bis-catechol siderophore to ampicillin or loracarbef can enhance the Minimal Inhibitory Concentration (MIC) against Gram-negative bacteria by over 8,000-fold compared to the unconjugated β-lactam [170].
- Dual Mechanisms: In Escherichia coli and Klebsiella pneumoniae, the enhanced activity was linked not only to improved uptake but also to significantly improved binding to PBPs. In contrast, for Pseudomonas aeruginosa and Acinetobacter baumannii, increased membrane permeability via TBDTs was the primary factor for improved activity [170].
- Iron-Dependent Activity: The activity of these conjugates is potentiated under iron-deficient conditions, which trigger the expression of TBDTs. Supplementing growth media with Fe³⁺ to a concentration of 0.1-1 μg/mL (approximately 1.8×10⁻⁶ to 1.8×10⁻⁵ M) can reverse this potentiation, restoring MIC values to those seen in standard media [170].
Table 1: Enhanced Efficacy of Siderophore-β-Lactam Conjugates (SID-βL)
| Conjugate | β-Lactam Component | Siderophore Type | Fold-Improvement in MIC | Key Mechanism |
|---|---|---|---|---|
| BAMP [170] | Ampicillin | Bis-catechol | >8,000-fold | Enhanced PBP binding (Enterobacterales) & Increased uptake [170] |
| BLOR [170] | Loracarbef | Bis-catechol | >8,000-fold | Enhanced PBP binding (Enterobacterales) & Increased uptake [170] |
| MCEF [170] | Cefaclor | Mixed bis-catechol-mono-hydroxamate | Data Specific | Increased uptake (A. baumannii, P. aeruginosa) [170] |
| Cefiderocol [170] | Ceftazidime/Cefepime derivative | Mono-chloro-catechol | Clinically effective vs. carbapenem-resistant strains | TBDT-mediated uptake [170] |
Bis-Beta-Lactam Antibiotics and Trimers
Another strategy involves creating molecules with multiple beta-lactam cores to enhance binding and evade resistance.
- Bis-Beta-Lactams: These are nearly symmetrical dimers, often derived from ampicillin or amoxicillin, designed to simultaneously bind two PBPs. This bifunctional nature increases their antibacterial potential, particularly against strains with mutated PBPs. For example, certain bis-beta-lactam compounds show significantly improved inhibitory concentration (IC₅₀) values for PBP1a mutation in E. coli compared to ampicillin alone [168].
- Trimer of Phenoxy-Methyl Penicillin: This structure exhibits beta-lactamase inhibitory activity. It demonstrates an IC₅₀ of 20 µg/mL (0.018 µmol/mL) against Enterobacter cloacae P99, which is comparable to the inhibitor sulbactam (IC₅₀ of 5 µg/mL or 0.02 µmol/mL) [168].
Table 2: Inhibitory Activity of Novel Multi-Core Beta-Lactam Structures
| Compound | Structure | Target / Assay | IC₅₀ / Activity | Comparison |
|---|---|---|---|---|
| Bis-beta-lactam (Compound III*) [168] | Dimer from Ampicillin | E. coli PBP1a mutation | 0.5 mcg/mL | Ampicillin: 13 mcg/mL [168] |
| Trimer of Phenoxy-Methyl Penicillin Sulphone [168] | Trimer | Enterobacter cloacae P99 (AmpC β-lactamase) | 20 µg/mL (0.018 µmol/mL) | Sulbactam: 5 µg/mL (0.02 µmol/mL) [168] |
Experimental Protocols for Key Assays
Protocol: Determination of Minimal Inhibitory Concentration (MIC) under Iron-Restricted Conditions
This protocol is critical for evaluating the efficacy of siderophore-antibiotic conjugates [170].
Media Preparation:
- Prepare standard cation-adjusted Mueller-Hinton Broth (MHBCA) as a control.
- Prepare Iron-Depleted Mueller-Hinton Broth (ID-MHBCA) by treating the broth with Chelex resin to remove cationic metals, including iron.
- Validate iron depletion by monitoring bacterial growth (OD₆₀₀) over 24 hours. Growth should be significantly impaired in ID-MHBCA compared to MHBCA.
- Prepare iron-supplemented ID-MHBCA by adding FeCl₃ to final concentrations of 0.1 µg/mL and 1 µg/mL to confirm that iron is the growth-limiting factor.
Inoculum Preparation:
- Grow fresh bacterial colonies (e.g., E. coli, P. aeruginosa, K. pneumoniae, A. baumannii) in MHBCA to a turbidity equivalent to a 0.5 McFarland standard.
- Dilute the suspension to achieve a final inoculum density of approximately 5 × 10⁵ CFU/mL in the test medium.
MIC Assay:
- Dispense the diluted bacterial inoculum into a 96-well microtiter plate.
- Perform two-fold serial dilutions of the test antibiotics (both conjugated and unconjugated) in the respective media (MHBCA, ID-MHBCA, and iron-supplemented ID-MHBCA).
- Include appropriate growth control and sterility control wells.
- Incubate the plates at 35°C for 16-20 hours.
- The MIC is defined as the lowest concentration of antibiotic that completely inhibits visible growth. The fold-improvement is calculated as (MIC of unconjugated β-lactam) / (MIC of conjugated β-lactam).
Protocol: Molecular Docking Analysis of Beta-Lactamase Inhibitors
This methodology is used to evaluate the potential of novel compounds as β-lactamase inhibitors, assessing their binding affinity and interactions [169].
Protein and Ligand Preparation:
- Retrieve 3D crystal structures of target β-lactamases (e.g., 5N5I, 7QPL, 1BLC) from the Protein Data Bank (PDB). Remove water molecules and co-crystallized ligands.
- Prepare the protein structures by adding hydrogen atoms and optimizing protonation states using molecular modeling software.
- Obtain or draw the 3D chemical structures of novel inhibitor candidates (e.g., L1, L2) and known inhibitors (e.g., clavulanic acid, avibactam, sulbactam). Perform energy minimization and assign appropriate charges.
Docking Simulation:
- Define the active site of the β-lactamase, typically around the catalytic serine for SBLs or the zinc ion cluster for MBLs.
- Use docking software (e.g., AutoDock Vina, GOLD) to perform flexible ligand docking into the rigid protein active site.
- Set the search space to encompass the entire active site cavity. Run multiple docking simulations for each ligand to generate a variety of potential binding poses.
Analysis of Results:
- The primary output is the binding affinity (estimated as kcal/mol). A more negative value indicates a stronger binding affinity.
- Analyze the top-ranking poses for key molecular interactions, such as hydrogen bonding, π-π stacking, and hydrophobic contacts, with active site residues.
- Compare the binding affinity and binding mode of novel inhibitors against established ones to predict relative efficacy.
Key stages in the research and development of novel beta-lactam conjugates and inhibitors, highlighting the role of computational modeling.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents for Beta-Lactam Stereochemistry and Efficacy Research
| Reagent / Material | Function / Application | Example Usage |
|---|---|---|
| Chelex-Treated Media [170] | Creates iron-depleted conditions to induce expression of bacterial iron-uptake systems (TBDTs) for evaluating sideromycin activity. | Used in MIC assays for siderophore-β-lactam conjugates like BAMP and MCEF [170]. |
| Recombinant Beta-Lactamases [171] [169] | Purified enzymes (Classes A, B, C, D) for in vitro inhibition assays and enzymatic degradation studies. | Determining IC₅₀ values of inhibitors; studying hydrolysis kinetics of novel β-lactams [169]. |
| Penicillin-Binding Proteins (PBPs) [170] | Purified bacterial PBPs for assessing the binding affinity and acylation efficiency of novel beta-lactams. | Bocillin FL fluorescence-based competitive binding assays to compare PBP affinity of conjugates vs. parent drugs [170]. |
| Cation-Adjusted Mueller-Hinton Broth (MHBCA) [170] | Standardized medium for antimicrobial susceptibility testing (AST) to ensure reproducible cation concentrations. | Performing reference MIC assays according to CLSI/EUCAST guidelines [170]. |
| Molecular Modeling & Docking Software [169] | Computational tools for predicting ligand-protein interactions and binding affinities prior to synthesis. | Screening novel β-lactamase inhibitors (e.g., L1, L2) against β-lactamase structures (e.g., PDB: 5N5I) [169]. |
| TonB-Dependent Transporter (TBDT) Mutants [170] [172] | Genetically engineered bacterial strains with deletions in specific siderophore receptors. | Elucidating the specific uptake pathways for siderophore-β-lactam conjugates [170]. |
Resistance Mechanisms and the Role of Target Modification
Despite advanced designs, bacteria evolve resistance. A key mechanism involves the modification of the antibiotic target, PBPs.
- PBP Mutations in P. aeruginosa: Mutations in the gene ftsI, which encodes PBP3, are a growing concern. Specific mutations like R504C/H and F533L are located within domains critical for the formation of the β-lactam-PBP3 inactivating complex. These mutations can lead to reduced affinity for beta-lactams, conferring resistance even to newer combination therapies [172].
- Altered Spectrum from Enzyme Mutations: Mutations in horizontally acquired beta-lactamases can alter their hydrolytic profile. For example, certain amino acid substitutions in the class A carbapenemase GES-1 (e.g., Gly170Ser) can shift its activity from an extended-spectrum β-lactamase (ESBL) to a carbapenem-hydrolyzing enzyme (GES-2, -5). Conversely, some mutations in KPC carbapenemases can paradoxically reduce carbapenem hydrolysis while increasing resistance to ceftazidime/avibactam, demonstrating a complex evolutionary landscape [172].
The strategic manipulation of beta-lactam stereochemistry is a powerful tool in the ongoing battle against antimicrobial resistance. The design of siderophore conjugates that exploit chiral recognition for targeted uptake, the synthesis of multi-core beta-lactams for enhanced PBP binding, and the development of novel beta-lactamase inhibitors all hinge on a deep understanding of three-dimensional molecular structure. Future research directions should include the large-scale in vivo validation of these promising compounds, the continued exploration of novel, broad-spectrum β-lactamase inhibitors capable of inhibiting metallo-β-lactamases (MBLs), and the application of advanced drug delivery systems. Integrating these sophisticated, stereochemistry-driven strategies is paramount for developing the next generation of effective beta-lactam antibiotics and ensuring their sustained clinical utility against multidrug-resistant pathogens.
Economic and Therapeutic Impact of Chiral Switches (e.g., Omeprazole to Esomeprazole)
Stereochemistry, the study of the three-dimensional structure of molecules, is a fundamental aspect of organic chemistry with profound implications in pharmaceutical research. Chirality, the property of molecules existing as non-superimposable mirror images (enantiomers), is particularly crucial as the human body is a chiral environment built from chiral building blocks like L-amino acids and D-sugars [173] [174]. This molecular asymmetry means that enantiomers can behave differently in biological systems, interacting selectively with chiral targets such as receptors, enzymes, and proteins [173].
The "chiral switch" strategy refers to the replacement on the market of a previously approved racemate (a 50:50 mixture of enantiomers) with its single enantiomer version [173] [174]. This process represents a significant trend in pharmaceutical development, sitting at the intersection of scientific innovation and commercial strategy. While chiral switches can offer genuine therapeutic advantages through improved pharmacokinetic and pharmacodynamic profiles, they also present a powerful evergreening tactic that allows manufacturers to extend market exclusivity for blockbuster drugs facing patent expiration [175] [176].
This review provides a comprehensive analysis of the economic and therapeutic implications of chiral switches, with a focused case study on the transition from omeprazole to esomeprazole. Through examination of clinical evidence, regulatory frameworks, and commercial outcomes, we aim to elucidate the complex balance between genuine therapeutic advancement and strategic market protection within the pharmaceutical industry.
Scientific Foundations of Chirality
Basic Principles and Terminology
- Enantiomers: Pairs of stereoisomers that are non-superimposable mirror images of one another, possessing identical physical and chemical properties in an achiral environment but potentially different biological activities [177].
- Racemate: A 1:1 mixture of two enantiomers, typically produced through traditional non-stereoselective chemical synthesis [176] [177].
- Eutomer and Distomer: The eutomer is the enantiomer with the desired, greater physiological activity, while the distomer is the other enantiomer, which may be inactive, less active, or contribute to adverse effects [176].
- Eudysmic Ratio: A quantitative measure of the difference in pharmacological activity between enantiomers, calculated as the ratio of the activity of the eutomer to that of the distomer [176].
The historical regulatory framework for chiral drugs was significantly shaped by the 1960s thalidomide tragedy, where one enantiomer provided therapeutic effects while the other caused severe birth defects [173] [174]. This event catalyzed stricter regulatory approaches, culminating in the 1992 FDA policy statement on developing stereoisomeric drugs, which encouraged the development of single-enantiomer products [173] [174].
Metabolic and Pharmacodynamic Considerations
In a chiral biological environment, enantiomers can exhibit stereoselectivity in their absorption, distribution, metabolism, and excretion (ADME) profiles [176]. For instance, enzymes in the cytochrome P450 system may metabolize enantiomers at different rates or through different pathways, leading to distinct pharmacokinetic profiles [176]. These differences can translate into clinical implications for therapeutic index, dosing regimens, and drug-drug interactions.
The Omeprazole to Esomeprazole Case Study
Chemical and Pharmacological Profiles
Omeprazole, a proton pump inhibitor (PPI), was initially marketed as a racemate under the brand name Prilosec. Its chirality stems from a sulfur atom in the sulfinyl group, creating two enantiomers: (R)-omeprazole and (S)-omeprazole [173]. As prodrugs, both enantiomers are activated in the acidic environment of parietal cells to form the same active sulfonamide metabolite that covalently binds to and inhibits the H+/K+-ATPase pump [177].
While the enantiomers are equipotent in their pharmacodynamic action at the proton pump, they exhibit stereoselective metabolism [177]. The (R)-enantiomer is predominantly metabolized by the CYP2C19 enzyme, which exhibits genetic polymorphism, leading to significant interindividual variability in its pharmacokinetics [175] [177]. In contrast, the (S)-enantiomer (esomeprazole) undergoes metabolism by multiple cytochrome P450 enzymes, resulting in a more consistent metabolic profile [175].
Table 1: Comparative Properties of Omeprazole and Esomeprazole
| Property | Omeprazole (Racemate) | Esomeprazole (S-enantiomer) |
|---|---|---|
| Chemical Composition | 50:50 mixture of R and S enantiomers | Pure S-enantiomer |
| Metabolic Pathway | Primarily CYP2C19 (polymorphic) | Multiple CYP enzymes |
| Systemic Exposure (AUC) | Reference | Approximately double that of the racemate at equal doses [178] |
| Interindividual Variability | High (7.5-fold in poor metabolizers) | Reduced (approximately 3-fold) [177] |
| FDA Approval Date | 1989 | 2001 |
Clinical Evidence and Therapeutic Equivalence
The clinical superiority of esomeprazole over omeprazole has been the subject of extensive debate and analysis. A 2015 systematic review and meta-analysis comparing the drugs at equivalent doses across 14 randomized controlled trials revealed nuanced findings [178]:
Table 2: Meta-Analysis of Efficacy Outcomes at Equivalent Doses [178]
| Therapeutic Context | Number of Studies | Odds Ratio (95% CI) | Statistical Significance |
|---|---|---|---|
| H. pylori Eradication (ITT) | 7 | 1.06 (0.83-1.36) | p = 0.63 |
| H. pylori Eradication (PP) | 7 | 1.07 (0.84-1.36) | p = 0.57 |
| GERD Symptom Resolution | 5 | 1.18 (1.01-1.38) | p = 0.04 |
| 24-hour Intragastric pH >4 | 3 | 1.57 (1.04-2.38) | p = 0.03 |
ITT: Intention-to-Treat; PP: Per-Protocol
The meta-analysis demonstrated that esomeprazole provided a statistically significant but clinically marginal benefit over omeprazole in GERD symptom resolution and intragastric pH control [178]. However, for H. pylori eradication as part of triple therapy, the drugs were therapeutically equivalent [178]. Importantly, many early industry-sponsored trials compared 40 mg esomeprazole to 20 mg omeprazole, potentially exaggerating perceived benefits due to dose disparity rather than enantiomeric superiority [178].
Experimental Methodology for Chiral Switch Evaluation
Research Objective: To compare the efficacy and pharmacokinetics of racemic omeprazole versus its single S-enantiomer (esomeprazole) at equivalent doses.
Study Design:
- Population: Adult patients (≥18 years) with confirmed GERD or requiring H. pylori eradication
- Intervention: Esomeprazole 20 mg or 40 mg once daily
- Comparator: Omeprazole 20 mg or 40 mg once daily
- Outcomes: Primary - healing rates of erosive esophagitis at 8 weeks; Secondary - 24-hour intragastric pH monitoring, symptom resolution scores, H. pylori eradication rates
Key Methodological Considerations:
- Utilize a randomized, double-blind, parallel-group design
- Employ crossover design for pharmacokinetic studies with adequate washout periods
- Implement 24-hour intragastric pH monitoring with standardized meals
- Analyze data by both intention-to-treat and per-protocol approaches
- Use HPLC with chiral columns for enantiomer-specific pharmacokinetic analysis
Economic and Regulatory Impact
Patent Strategy and Market Exclusivity
AstraZeneca deployed sophisticated intellectual property strategies to protect their blockbuster drug omeprazole, whose patent was set to expire in 2001 [175]. The company created a "patent thicket" - in the US, omeprazole was protected by 40 patents, while in Australia, 61 additional patents were filed [175]. This strategy effectively delayed generic competition and created a protective window for the launch of esomeprazole in 2001, just months before omeprazole's patent expiration [175].
The chiral switch successfully transferred consumer loyalty from Omeprazole (Prilosec) to Esomeprazole (Nexium), allowing AstraZeneca to maintain monopoly pricing [175]. At its peak, omeprazole was the world's best-selling drug with annual US sales of $6 billion, and by 2010, esomeprazole became AstraZeneca's bestseller with US sales of $5.63 billion, effectively compensating for the plummeting sales of omeprazole [175].
Evergreening and Economic Implications
The omeprazole-esomeprazole case exemplifies "evergreening" - the use of legal, commercial, and technological strategies to extend the commercial exclusivity of successful products [175]. This practice has significant economic consequences:
- Increased financial burden: Patients and payers face higher costs for the patented enantiomer versus generic racemate
- Potential impact on medication access: Higher prices may limit access for some patient populations
- Market distortion: The pure enantiomer quickly absorbs market share from the racemic precursor, redirecting use from generic versions [173]
Regulatory pathways facilitate chiral switches, as FDA and EMA cannot legally require active comparators in clinical trials for drug approval [173]. Consequently, pure enantiomer products can enter the market by demonstrating superiority over placebo rather than their precursor racemate [173], creating a lower evidentiary bar for approval.
Visualization of Chiral Switch Development
Diagram 1: Chiral Switch Development Workflow. This diagram illustrates the strategic development pathway from a racemic drug facing patent expiry to a market-protected single enantiomer product.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents for Chiral Switch Research
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Chiral HPLC Columns | Enantiomer separation and quantification | Cellulose/amylose-based stationary phases (Chiralpak, Chiralcel) |
| Enantioselective Synthesis Catalysts | Asymmetric synthesis of single enantiomers | Chiral Lewis acids, BINAP-metal complexes, Jacobsen's catalyst |
| Cytochrome P450 Enzymes | Metabolic profiling of enantiomers | Recombinant CYP2C19, CYP3A4, CYP2C9 isoforms |
| Animal Disease Models | In vivo efficacy assessment | Rat gastric ulcer models, GERD animal models |
| pH Monitoring Systems | Pharmacodynamic assessment | 24-hour intragastric pH monitoring equipment |
| Crystallization Resolving Agents | Classical enantiomer separation | Diastereomeric salt formation with chiral acids/bases |
The chiral switch from omeprazole to esomeprazole represents a paradigm for understanding the complex interplay between scientific innovation and commercial strategy in pharmaceutical development. From a therapeutic perspective, the transition offered modest clinical benefits primarily through improved pharmacokinetic profiles rather than enhanced pharmacodynamic activity [177] [178]. The marginal superiority in GERD treatment must be weighed against significantly higher costs [178].
From an economic standpoint, the strategy proved enormously successful for AstraZeneca, allowing the company to maintain market dominance in the PPI category through sophisticated patent strategies and marketing [175]. However, this approach raises important questions about the appropriate balance between innovation reward and public health interests, particularly when the therapeutic advantages of a chiral switch are minimal [175].
The future of chiral switches will likely be shaped by evolving regulatory landscapes, greater scrutiny from payers regarding cost-effectiveness, and advances in enantioselective synthesis technologies. For researchers and drug development professionals, critical evaluation of both therapeutic merits and commercial implications remains essential when assessing the value of chiral switch strategies in pharmaceutical development.
Conclusion
Stereochemistry represents a fundamental determinant of pharmaceutical behavior that directly impacts drug safety, efficacy, and development success. The integration of robust stereochemical principles with advanced analytical methodologies enables precise characterization of chiral drug molecules, while comprehensive understanding of isomer-specific biological activities informs rational drug design. Future directions in stereochemistry research will likely focus on developing more efficient chiral separation technologies, computational prediction models for stereochemical outcomes, and personalized medicine approaches that account for individual metabolic variations in enantiomer processing. The continued emphasis on stereoselective analysis throughout the drug development pipeline will remain essential for delivering safer, more effective therapeutics to market while meeting evolving regulatory standards for chiral drug evaluation.
References
- 1. Stereochemistry - (Inorganic Chemistry I) [https://fiveable.me/key-terms/inorganic-chemistry-i/stereochemistry]
- 2. Stereochemistry [https://www.geeksforgeeks.org/chemistry/stereochemistry/]
- 3. Stereochemistry in Drug Action - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC353039/]
- 4. Predicting the stereoselectivity of chemical reactions by ... [https://www.nature.com/articles/s41598-024-62158-0]
- 5. D- and L- Notation For Sugars [https://www.masterorganicchemistry.com/2017/05/24/d-and-l-sugars/]
- 6. Quantification of Stereochemical Communication in Metal– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5012202/]
- 7. Part 9: Stereochemistry in Drug Discovery and Development [https://chiralpedia.com/blog/part-9-stereochemistry-in-drug-discovery-and-development/]
- 8. Structural Isomerism in Organic Molecules [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Fundamentals/Isomerism_in_Organic_Compounds/Structural_Isomerism_in_Organic_Molecules]
- 9. The Influence of Geometric Isomers on Drug Delivery ... [https://eureka.patsnap.com/report-the-influence-of-geometric-isomers-on-drug-delivery-systems]
- 10. 2.7: Isomerism Introduction [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Wade)_Complete_and_Semesters_I_and_II/Map%3A_Organic_Chemistry_(Wade)/02%3A_Structure_and_Properties_of_Organic_Molecules/2.07%3A_Isomerism_Introduction]
- 11. Types of Isomers: Constitutional, Stereoisomers, ... [https://www.masterorganicchemistry.com/2018/09/10/types-of-isomers/]
- 12. 5.1 Overview of Isomerism and Stereoisomers - Chad's Prep [https://www.chadsprep.com/chads-organic-chemistry-videos/stereoisomers/?srsltid=AfmBOoqTZ6d5mr7hR6yU3mP0SyuRBCnpJbPtRzgaoP-lpx-2T8sPJ3cv]
- 13. Structural isomer [https://en.wikipedia.org/wiki/Structural_isomer]
- 14. Stereoisomerism [https://en.wikipedia.org/wiki/Stereoisomerism]
- 15. structural isomerism and stereoisomerism [https://www.chemistrystudent.com/cie-a-level/13-organic/structural-isomerism-and-stereoisomerism.html]
- 16. Stereoisomerism | Definition, Examples, Types, & Chirality [https://www.britannica.com/science/stereoisomerism]
- 17. Exploring the Potential of Pyridine Carboxylic Acid Isomers ... [https://pubmed.ncbi.nlm.nih.gov/40420948/]
- 18. Part 1: Introduction to Stereochemistry [https://chiralpedia.com/blog/part-1-introduction-to-stereochemistry/]
- 19. Chiral Drugs: An Overview - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3614593/]
- 20. Catalytic asymmetric constructions of nitrogen, boron and ... [https://www.nature.com/articles/s41467-025-64905-x]
- 21. Chirality shock: Geneva chemists forge millennia-stable ' ... [https://www.sciencedaily.com/releases/2025/07/250715043355.htm]
- 22. New class of chiral molecules offers strong stability for drug ... [https://www.drugtargetreview.com/news/178148/new-class-of-chiral-molecules-offers-strong-stability-for-drug-development/]
- 23. Stereochemistry-aware string-based molecular generation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582147/]
- 24. Catalytic Light-induced Deracemization (CALIDE) [https://www.ch.nat.tum.de/en/oc1/research/calide/]
- 25. Stereochemical diversity as a source of discovery in ... [https://www.sciencedirect.com/science/article/pii/S2666246922000106]
- 26. 8.1: Types of Isomers [https://chem.libretexts.org/Courses/Thompson_Rivers_University/CHEM_1500%3A_Chemical_Bonding_and_Organic_Chemistry/08%3A_Organic_Chemistry_II_-_Stereochemistry/8.01%3A_Types_of_Isomers]
- 27. Enantioselectivity in Drug Pharmacokinetics and Toxicity [https://pmc.ncbi.nlm.nih.gov/articles/PMC8197169/]
- 28. The Significance of Chirality in Drug Design and Development [https://pmc.ncbi.nlm.nih.gov/articles/PMC5765859/]
- 29. Enantiomers vs Diastereomers vs The Same? Two ... [https://www.masterorganicchemistry.com/2019/03/08/enantiomers-diastereomers-or-the-same-1-using-models/]
- 30. Difference Between Enantiomers and Diastereomers with ... [https://www.vedantu.com/jee-main/chemistry-difference-between-enantiomers-and-diastereomers]
- 31. Difference Between Enantiomers And Diastereomers [https://byjus.com/chemistry/difference-between-enantiomers-and-diastereomers/]
- 32. A Holistic Approach to Determining Stereochemistry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8745598/]
- 33. 5.4: Pasteur's Discovery of Enantiomers [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(OpenStax)/05%3A_Stereochemistry_at_Tetrahedral_Centers/5.04%3A_Pasteur's_Discovery_of_Enantiomers]
- 34. Pasteur and chirality: A story of how serendipity favors the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9291139/]
- 35. Architecture of the molecules of life, a contribution of Louis ... [https://comptes-rendus.academie-sciences.fr/chimie/item/CRCHIM_2020__23_1_3_0/]
- 36. molecular structures - Pasteurized Crystals – Tartaric acid [https://www.iycr2014.org/learn/crystallography365/categories/molecular-structures?result_105799_result_page=101]
- 37. Who Discovered Stereochemistry? | The Science Blog [https://www.reagent.co.uk/blog/who-discovered-stereochemistry/]
- 38. 5.7: Meso Compounds [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/05%3A_Stereochemistry_at_Tetrahedral_Centers/5.07%3A_Meso_Compounds]
- 39. Meso compound [https://en.wikipedia.org/wiki/Meso_compound]
- 40. Meso Compounds [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Chirality/Meso_Compounds]
- 41. 5.7 Meso Compounds – Organic Chemistry: A Tenth Edition [https://ncstate.pressbooks.pub/organicchem/chapter/meso-compounds/]
- 42. 5.7 Meso Compounds - Organic Chemistry [https://fiveable.me/organic-chem/unit-5/meso-compounds/study-guide/2SaU6urswCz6jzF0]
- 43. Meso Compounds [https://www.chemistrysteps.com/symmetry-chiralty-meso-compunds/]
- 44. The Meso Trap [https://www.masterorganicchemistry.com/2011/01/12/the-meso-trap/]
- 45. 5.1 Overview of Isomerism and Stereoisomers - Chad's Prep [https://www.chadsprep.com/chads-organic-chemistry-videos/stereoisomers/?srsltid=AfmBOopKz7rsgrr-7Jgyk7QqUWKqFThe8ZYnBhnQ7B2IENB_XsSjKx7_]
- 46. Stereochemistry [https://en.wikipedia.org/wiki/Stereochemistry]
- 47. Chirality and Stereoisomers [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Chirality/Chirality_and_Stereoisomers]
- 48. Thalidomide [https://www.acs.org/molecule-of-the-week/archive/t/thalidomide.html]
- 49. The Rise, Fall and Subsequent Triumph of Thalidomide [https://pmc.ncbi.nlm.nih.gov/articles/PMC3573415/]
- 50. How a courageous physician-scientist saved the U.S. from ... [https://www.uchicagomedicine.org/forefront/biological-sciences-articles/2020/march/courageous-physician-scientist-saved-the-us-from-a-birth-defects-catastrophe]
- 51. Death and Rebirth of the Thalidomide Molecule [https://pmc.ncbi.nlm.nih.gov/articles/PMC7936918/]
- 52. Understanding the Thalidomide Chirality in Biological ... [https://www.nature.com/articles/s41598-018-35457-6]
- 53. After 60 years, scientists uncover how thalidomide ... [https://www.dana-farber.org/newsroom/news-releases/2018/after-60-years-scientists-uncover-how-thalidomide-produced-birth-defects]
- 54. 5.9: A Review of Isomerism [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/05%3A_Stereochemistry_at_Tetrahedral_Centers/5.09%3A_A_Review_of_Isomerism]
- 55. Introduction to Assigning (R) and (S): The Cahn-Ingold- ... [https://www.masterorganicchemistry.com/2016/10/20/introduction-to-assigning-r-and-s-the-cahn-ingold-prelog-rules/]
- 56. Part 3: Nomenclature and Configuration [https://chiralpedia.com/blog/part-3-nomenclature-and-configuration/]
- 57. Cahn–Ingold–Prelog priority rules [https://en.wikipedia.org/wiki/Cahn%E2%80%93Ingold%E2%80%93Prelog_priority_rules]
- 58. 8.4: Absolute Configuration- R-S Sequence Rules [https://chem.libretexts.org/Courses/Thompson_Rivers_University/CHEM_1500%3A_Chemical_Bonding_and_Organic_Chemistry/08%3A_Organic_Chemistry_II_-_Stereochemistry/8.04%3A_Absolute_Configuration-_R-S__Sequence_Rules]
- 59. CIP (Cahn-Ingold-Prelog) Priorities [https://learn.openochem.org/learn/first-semester-topics/stereochemistry/cip-cahn-ingold-prelog-priorities-and-configuration]
- 60. 3.3: Configurations of Chiral Molecules- the Cahn-Ingold- ... [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/OCLUE%3A_Organic_Chemistry_Life_the_Universe_and_Everything_(Copper_and_Klymkowsky)/03%3A_Conformations_and_Configurations_-_the_consequences_of_the_three-dimensional_nature_of_carbon_compounds/3.03%3A_Configurations_of_Chiral_Molecules-_the_Cahn-Ingold-Prelog_Convention]
- 61. Absolute Configurations Assigning R and S - Chad's Prep® [https://www.chadsprep.com/chads-organic-chemistry-videos/absolute-configurations/?srsltid=AfmBOoo_tZrtOECm7cUkgxRkVgiUmfTMVqWykoeoXR_BMBGi5OzsHABm]
- 62. A review of stereochemical implications in the generation of ... [https://pubs.rsc.org/en/content/articlehtml/2016/em/c6em00354k]
- 63. Computational Approaches and Use of Chiroptical Probes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8945943/]
- 64. Complementarity of electronic and vibrational circular ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993615002071]
- 65. Applications of chiroptical spectroscopy for the ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708598000612]
- 66. Impact of conformation and intramolecular interactions on ... [https://www.nature.com/articles/s42004-023-00944-z]
- 67. Chiroptical Spectroscopy, Theoretical Calculations, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11858395/]
- 68. Chromatography and Crystallization in Chiral Resolution [https://chiralpedia.com/blog/breaking-down-the-methods-chromatography-and-crystallization-in-chiral-resolution/]
- 69. A review of drug isomerism and its significance - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3678675/]
- 70. Enantiomer Identification (HPLC/SFC) [https://www.solutions.bocsci.com/enantiomer-identification-hplc-scf.htm]
- 71. Finding the Best Separation for Enantiomeric Mixtures [https://www.chromatographyonline.com/view/finding-best-separation-enantiomeric-mixtures]
- 72. Chromatographic isolation for performing enantiomer-specific ... [https://progearthplanetsci.springeropen.com/articles/10.1186/s40645-025-00764-w]
- 73. High Efficiency Chiral Separations in HPLC and SFC [https://www.chromatographyonline.com/view/high-efficiency-chiral-separations-hplc-and-sfc]
- 74. The Science and Strategy Behind Isomer Separation [https://rotachrom.com/the-science-and-strategy-behind-isomer-separation-advancements-in-chromatography-for-precision-purification-isomerism-part-1/]
- 75. Preparative Supercritical Fluid Chromatography for Chiral ... [https://selvita.com/our-science/resources/blog-articles/faster-greener-and-more-efficient-preparative-supercritical-fluid-chromatography-for-chiral-separations]
- 76. HPLC Update Day 2: Modern Method Development [https://www.chromatographyonline.com/view/hplc-update-day-2-modern-method-development]
- 77. Asymmetric Synthesis of US-FDA Approved Drugs over Five ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10052577/]
- 78. Understanding the Fundamentals of Asymmetric Synthesis [https://chiralpedia.com/blog/understanding-the-fundamentals-of-asymmetric-synthesis/]
- 79. Global Market to Surpass $10 Billion by 2030 [https://www.businesswire.com/news/home/20250422489286/en/Chiral-Technology-Research-Business-Report-2025-Global-Market-to-Surpass-%2410-Billion-by-2030---Demand-for-Purity-in-Pharma-Ingredients-Strengthens-Business-Case-for-Chiral-Methods---ResearchAndMarkets.com]
- 80. 5.9 A Review of Isomerism - Organic Chemistry [https://fiveable.me/organic-chem/unit-5/review-isomerism/study-guide/fUd9POrpbZcc7rA0]
- 81. Chiral Separation and Enantiomeric Analysis: Critical ... [https://www.americanpharmaceuticalreview.com/Featured-Articles/619871-Chiral-Separation-and-Enantiomeric-Analysis-Critical-Importance-in-Pharmaceutical-Development/]
- 82. Catalyzed Enantioselective Organic Synthesis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550611/]
- 83. Significance and challenges of stereoselectivity assessing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5762452/]
- 84. Connecting the complexity of stereoselective synthesis to the ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc07461k]
- 85. Mechanism and origin of stereoselectivity and regioselectivity ... [https://pubs.rsc.org/en/content/articlehtml/2025/cy/d5cy00519a]
- 86. Impact of stereochemistry in 3D energetic materials science [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc02800k]
- 87. Isomer analysis by mass spectrometry in clinical science [https://www.sciencedirect.com/science/article/abs/pii/S0165993622003909]
- 88. 5.1 Overview of Isomerism and Stereoisomers - Chad's Prep [https://www.chadsprep.com/chads-organic-chemistry-videos/stereoisomers/?srsltid=AfmBOooBdm0qRQvNtMErUtYOwziWMYEOl-MO_Kiu5dRsvAtTvkTCRhm2]
- 89. Development and validation of a chiral bioanalytical ... [https://www.sciencedirect.com/science/article/pii/S1573412925000635]
- 90. Quantitative analysis of lycopene isomers in human plasma ... [https://pubmed.ncbi.nlm.nih.gov/12622371/]
- 91. The integration of LC-MS and NMR for the analysis of low ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6339611/]
- 92. Bioanalytical method validation: An updated review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658022/]
- 93. Clinical Questions: Are Single Enantiomers Always Better? [https://chiralpedia.com/blog/episode-7-clinical-questions-are-single-enantiomers-always-better/]
- 94. Racemic drugs are not necessarily less efficacious and ... [https://www.sciencedirect.com/science/article/pii/S0928098725000818]
- 95. Racemic drugs are not necessarily less efficacious and ... [https://pubmed.ncbi.nlm.nih.gov/40154891/]
- 96. Chiral Drugs [https://www.galchimia.com/chiral-drugs/]
- 97. pure enantiomers of drugs instead of racemic mixtures]. ... [https://kclpure.kcl.ac.uk/portal/en/publications/chiral-switch-pure-enantiomers-of-drugs-instead-of-racemic-mixtur/]
- 98. A discussion of strychnine and α-methylene-γ-butyrolactone [https://www.sciencedirect.com/science/article/pii/S1090780715002402]
- 99. Identifying the Conformational Isomers of Single-Molecule ... [https://www.sciencedirect.com/science/article/pii/S2451929420303740]
- 100. an approach for analyzing the conformational behaviour ... [https://pubmed.ncbi.nlm.nih.gov/9785960/]
- 101. GEOM, energy-annotated molecular conformations for ... [https://www.nature.com/articles/s41597-022-01288-4]
- 102. Conformational energies of reference organic molecules [https://link.springer.com/article/10.1007/s10822-023-00513-5]
- 103. Configurational assignments of type-I polyketide synthase ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d4np00061g]
- 104. Advances in chiral analysis: from classical methods to ... [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d4cs01202j]
- 105. 5.9 A Review of Isomerism - Organic Chemistry [https://openstax.org/books/organic-chemistry/pages/5-9-a-review-of-isomerism]
- 106. Unconventional approaches for chiral resolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11180637/]
- 107. Dynamic radical recombination enabling stereodivergent ... [https://www.nature.com/articles/s41467-025-64491-y]
- 108. Recent advances on asymmetric reduction via dynamic ... [https://www.sciencedirect.com/science/article/abs/pii/S1001841725011076]
- 109. Chiral resolution [https://en.wikipedia.org/wiki/Chiral_resolution]
- 110. Unconventional Approaches in Chiral Resolution [https://chiralpedia.com/blog/unconventional-approaches-in-chiral-resolution-new-horizons/]
- 111. Machine Learning Classification of Chirality and Optical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12076508/]
- 112. Advances and unresolved challenges in the structural ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269716303037]
- 113. Biological Matrix Effects in Quantitative Tandem Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4695332/]
- 114. Separation of Cis-Trans Phospholipid Isomers using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3397781/]
- 115. Strategies for the Detection and Elimination of Matrix ... [https://www.chromatographyonline.com/view/strategies-detection-and-elimination-matrix-effects-quantitative-lc-ms-analysis]
- 116. New techniques of on-line biological sample processing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5071623/]
- 117. Metabolite identification and quantitation in LC-MS/MS-based metabolomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3278153/]
- 118. Identification of Metabolite Interference Is Necessary for Accurate LC-MS Targeted Metabolomics Analysis - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10209981/]
- 119. Role of Stereochemistry on the Biological Activity of Nature ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10095986/]
- 120. LC-MS-based metabolomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3699692/]
- 121. August 18-19, 2025 | Valencia - Synthetic Organic Chemistry [https://organic.chemistryconferences.org/events-list/stereochemistry]
- 122. Using ammonium hydroxide to improve peptide epimer ... [https://pubmed.ncbi.nlm.nih.gov/41136006/]
- 123. Recent developments in the high-throughput separation of ... [https://www.sciencedirect.com/science/article/pii/S0731708523005630]
- 124. HPLC 2025 Preview: Boosting the Separation Power of LC ... [https://www.chromatographyonline.com/view/boosting-the-separation-power-of-lc-lc]
- 125. Enhancing pyrrolizidine alkaloid separation and detection [https://www.sciencedirect.com/science/article/abs/pii/S0021967325002110]
- 126. The Chiral Switch: A Pharmaceutical Tactic to Prolong ... [https://www.drugpatentwatch.com/blog/the-chiral-switch-a-pharmaceutical-tactic-to-prolong-exclusivity/?srsltid=AfmBOoqN_nP-qAWuPRdWiLt1_ghTF6cCScPKNNmkNp_ClWWsvcDnyPQO]
- 127. Part 10: Stereochemistry in Pharmaceutical Sciences [https://chiralpedia.com/blog/part-10-current-trends-and-future-directions/]
- 128. Stereochemistry Market Current & Forecast Sizing Trend [https://htfmarketinsights.com/report/4387171-stereochemistry-market]
- 129. The cost benefit ratio of enantiomeric drugs [https://pubmed.ncbi.nlm.nih.gov/7588989/]
- 130. The cost benefit ratio of enantiomeric drugs [https://link.springer.com/article/10.1007/BF03192284]
- 131. The Dark Art of Chemistry - Chiral Chromatography [https://www.sygnaturediscovery.com/news-and-events/blog/the-dark-art-of-chemistry-chiral-chromatography/]
- 132. Chirality of New Drug Approvals (2013–2022) - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC10895675/]
- 133. Chiral Bioequivalence – An Explainer [https://chiralpedia.com/blog/chiral-bioequivalence-an-explainer/]
- 134. Validation and Verification of Analytical Testing Methods ... [https://www.federalregister.gov/documents/2025/01/07/2024-31541/validation-and-verification-of-analytical-testing-methods-used-for-tobacco-products-guidance-for]
- 135. Chiral analysis [https://en.wikipedia.org/wiki/Chiral_analysis]
- 136. Chiral HPLC Column [https://www.phenomenex.com/techniques/hplc-chiral?srsltid=AfmBOooJUD2joVvdOXzIk90Qe-QqUXEvxLhn9aMNGVd74WvaSfVgwhZ9]
- 137. A Holistic Approach to Determining Stereochemistry of ... [https://www.mdpi.com/1422-0067/23/1/273]
- 138. How to Determine the R and S Configuration [https://www.chemistrysteps.com/how-to-determine-r-and-s-configuration/]
- 139. Stereochemistry and Validation of Macromolecular Structures [https://pmc.ncbi.nlm.nih.gov/articles/PMC5560084/]
- 140. Comparison of X-ray Crystallography, NMR and EM [https://www.creative-biostructure.com/comparison-of-crystallography-nmr-and-em_6.htm?srsltid=AfmBOooPpObbGIWGDoQ3LegTfrrL1VvWdvZdQrYnOo5GROXmqNRqs7M8]
- 141. Validation of X-ray Crystal Structure Ensemble ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10270724/]
- 142. How accurate is circular dichroism-based model validation? [https://link.springer.com/article/10.1007/s00249-020-01457-6]
- 143. Core packing of well‐defined X‐ray and NMR structures is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9277709/]
- 144. Stereochemistry And Conformational Analysis | Organic ... [https://scitechseries.com/organic-chemistry/program/scientific-sessions/stereochemistry-and-conformational-analysis]
- 145. Part 4: Stereochemistry in Drug Action and Pharmacology [https://chiralpedia.com/blog/part-4-stereochemistry-in-drug-action-and-pharmacology/]
- 146. NE-ADME 2025 Agenda [https://www.bostonsociety.org/NE-ADME/agenda.html]
- 147. A machine learning approach to population ... [https://www.nature.com/articles/s43856-025-01054-8]
- 148. Advancing drug development with “Fit-for-Purpose” modeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12436579/]
- 149. Prediction of pharmacokinetic/pharmacodynamic ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1578117/full]
- 150. Insights of affinity-based probes for target identification in ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425004763]
- 151. Top 6 Visualizations for Quantitative Data Analysis Methods [https://chartexpo.com/blog/quantitative-data-analysis-methods]
- 152. Rules of engagement: Determinants of chemokine receptor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12556802/]
- 153. Development of New Stereoisomeric Drugs May 1992 [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs]
- 154. Stereoisomer-specific bacterial mechanisms for ... [https://www.sciencedirect.com/science/article/abs/pii/S0304389425005035]
- 155. Enzymatic twists evolved stereo-divergent alkaloids in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12177067/]
- 156. Stereospecific lactylation in bacteriology: L/D-lactate ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1693700/full]
- 157. Quantitative Comparison of Statistical Methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9227835/]
- 158. Therapeutic index [https://en.wikipedia.org/wiki/Therapeutic_index]
- 159. Therapeutic Index [https://pharmacologycanada.org/Therapeutic-Index]
- 160. Comparative therapeutic index, lethal time and safety margin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7296648/]
- 161. Therapeutic index, ED50, TD50 and LD50 [https://derangedphysiology.com/main/cicm-primary-exam/pharmacodynamics/Chapter-413/therapeutic-index-ed50-td50-and-ld50]
- 162. Drug Therapeutic Index - an overview [https://www.sciencedirect.com/topics/nursing-and-health-professions/drug-therapeutic-index]
- 163. Drug toxicity prediction based on genotype-phenotype ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12597050/]
- 164. Table of Substrates, Inhibitors and Inducers [https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers]
- 165. Tox-Scapes: A visual and quantitative tool for selecting ... [https://www.sciencedirect.com/science/article/abs/pii/S0959652625013204]
- 166. β-Lactamases and β-Lactamase Inhibitors in the 21st Century [https://www.sciencedirect.com/science/article/pii/S0022283619301822]
- 167. Organic Chemistry Stereoisomers. Free In-Depth Study ... [https://chemistry.coach/organic-chemistry-1/stereoisomers]
- 168. Promising Future of Novel Beta-Lactam Antibiotics Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12517300/]
- 169. Discovery and Analysis of Two Novel β-Lactamase Inhibitors [https://www.sciencedirect.com/science/article/abs/pii/S0019452225005709]
- 170. Unraveling the mechanisms behind the enhanced efficacy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12592502/]
- 171. Biochemical exploration of β-lactamase inhibitors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9888030/]
- 172. Emerging resistance mechanisms to newer β-lactams in ... [https://www.sciencedirect.com/science/article/abs/pii/S1198743X25001314]
- 173. Chiral Switch: Between Therapeutical Benefit and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8877306/]
- 174. Chiral Switch: Between Therapeutical Benefit and ... [https://www.mdpi.com/1424-8247/15/2/240]
- 175. A case study of AstraZeneca's omeprazole/esomeprazole ... [https://gabi-journal.net/a-case-study-of-astrazenecas-omeprazole-esomeprazole-chiral-switch-strategy.html]
- 176. The Chiral Switch: A Pharmaceutical Tactic to Prolong ... [https://www.drugpatentwatch.com/blog/the-chiral-switch-a-pharmaceutical-tactic-to-prolong-exclusivity/?srsltid=AfmBOoqlNpU5MrY7_2WNObM-DZVNmnQAIJVR_zb0FaR-UgKjpgdo7Op1]
- 177. Inside the isomers: the tale of chiral switches [https://australianprescriber.tg.org.au/articles/inside-the-isomers-the-tale-of-chiral-switches-1.html]
- 178. Comparative efficacy of esomeprazole and omeprazole [https://pmc.ncbi.nlm.nih.gov/articles/PMC4647708/]